Expert médical de l'article

Nouvelles publications
Hamartoma
Dernière revue: 29.06.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
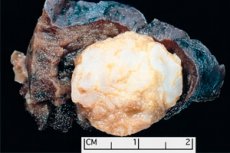
Une formation de type tumoral localisée dans n'importe quelle région anatomique résultant d'une croissance anormale de tissu bénin, est définie en médecine comme un hamartome (du grec hamartia - erreur, défaut). [ 1 ]
Épidémiologie
Statistiquement, les hamartomes représentent 1,2 % des néoplasies bénignes. Leur prévalence est estimée à environ 0,25 % de la population générale et peut atteindre 8 % de l'ensemble des néoplasies pulmonaires. La plupart des hamartomes pulmonaires sont diagnostiqués fortuitement chez des patients âgés de 40 à 70 ans, mais ils sont très rares en pédiatrie.
En général, la plupart des hamartomes sont diagnostiqués chez les hommes, bien que dans le rein, ils soient plus fréquents chez les femmes et soient identifiés à l'âge moyen.
Environ 5 % des tumeurs bénignes du sein sont des hamartomes et touchent le plus souvent les femmes de plus de 35 ans.
80 à 90 % des lésions hamartomateuses du cerveau et plus de 50 % des hamartomes du cœur sont associés à une sclérose tubéreuse.
Causes ng hamartomas
Les gamartomes sont des malformations congénitales bénignes, formées à partir de tissus mésenchymateux provenant des feuillets germinaux. Leur apparition est due à une division cellulaire incontrôlée de tissus cytologiquement normaux (conjonctif, muscle lisse, graisse ou cartilage), caractéristiques d'une localisation anatomique donnée, et à leur prolifération focale lors de l'embryogenèse de presque tous les organes et structures anatomiques.
L’apparition de plusieurs hamartomes chez le même patient est souvent appelée hamartomatose ou hamartome pléiotrope.
Ces tumeurs peuvent survenir de manière sporadique ou en présence de certaines maladies héréditaires autosomiques dominantes ainsi que de syndromes génétiquement déterminés.
Dans de nombreux cas, les hamartomes se forment lorsqu'une maladie génétique rare de nature multisystémique - la sclérose tubéreuse - se manifeste peu après la naissance, ou dans la maladie familiale de Recklinghausen - la neurofibromatose de type 1. [ 2 ]
Facteurs de risque
Les principaux facteurs de risque de formation d'hamartomes comprennent la présence de syndromes génétiques de polypose hamartomateuse dans les antécédents des patients, notamment:
- Syndrome d'hamartomes multiples - syndrome de Cowden, dans lequel se forment de multiples hamartomes d'origine ecto-, ento- et mésodermique, une polypose gastro-intestinale et des manifestations mucocutanées sont observées;
- Syndrome de Peutz-Jeghers-Turen (caractérisé par le développement de polypes hamartomateux bénins dans le tractus gastro-intestinal);
- syndrome de Protée;
- Syndrome de Weil - polypose juvénile du côlon;
- Syndrome de Bannayan-Riley-Ruvalcaba, qui, comme le syndrome de Cowden, produit de multiples hamartomes (polypes hamartomateux) de l'intestin;
- Syndrome de Carney-Stratakis et complexe de Carney.
De plus, des hamartomes se forment chez les patients atteints du syndrome de Watson héréditaire et dans les cas de syndrome de Pallister-Hall sporadique ou congénital avec hamartome hypothalamique et polydactylie.
Pathogénèse
Le mécanisme de prolifération accrue des tissus germinaux avec formation de malformations de type tumoral dans divers organes s'explique par des aberrations chromosomiques et des mutations génétiques qui peuvent survenir spontanément ou être héréditaires.
Dans la sclérose tubéreuse, des mutations des gènes TSC1 ou TSC2, suppresseurs de tumeurs qui préviennent et inhibent la prolifération excessive (croissance et division cellulaires trop rapides ou incontrôlées), ont été identifiées. Dans la neurofibromatose de type 1 et le syndrome de Watson, des mutations germinales du gène suppresseur de tumeur mitochondrial NF1 ont également été identifiées.
Dans le syndrome tumoral d'hamartome, qui combine les syndromes de Cowden, de Protea, de Bannayan-Riley-Ruvalcaba et de polypose juvénile, la pathogenèse est associée à une mutation du gène PTEN, qui code une enzyme impliquée dans la régulation de la prolifération et est considéré comme un gène suppresseur de tumeur.
Des mutations du gène STK11 codant la structure et la fonction de l'une des enzymes sérines transmembranaires, réduisant sa capacité à freiner la division cellulaire, conduisent au syndrome de Peutz-Jeghers-Turen, avec développement de polypes intestinaux et de lésions cutanées pigmentées. Une mutation du gène GLI3, un facteur de transcription impliqué dans la formation des tissus intra-utérins, a été identifiée dans le syndrome de Pallister-Hall.
Ainsi, la croissance cellulaire incontrôlée due à des mutations génétiques conduit à la formation d'hamartomes.
Symptômes ng hamartomas
En fonction de la localisation des hamartomes, leurs types sont distingués et chacun d'eux a sa propre structure et sa propre symptomatologie.
Hamartome du poumon
L'hamartome pulmonaire peut se former dans n'importe quel lobe et dans les parties périphériques des poumons. Il est constitué de tissus pulmonaires normaux: adipeux, épithéliaux, fibreux et cartilagineux. Dans 80 % des cas, la composante chondroïde (cellules cartilagineuses hyalines) prédomine, avec l'inclusion d'adipocytes (cellules du tissu adipeux) et de cellules épithéliales des voies respiratoires. [ 3 ]
Les noms antérieurs: hamartome chondroïde, mésenchymome, hamartome chondromateux ou hamartochondrome ne sont actuellement pas recommandés par l’OMS.
L'hamartome kystique mésenchymateux du poumon, en revanche, est moins fréquent et est associé au syndrome de Cowden chez la plupart des patients.
La lésion hamartomateuse du poumon peut ne pas se manifester, mais peut provoquer des symptômes sous forme de toux chronique (souvent avec hémoptysie), de respiration sifflante et de difficultés respiratoires. [ 4 ]
Un hamartome du cœur
Les tumeurs cardiaques primaires bénignes chez l'adulte comprennent l'hamartome myocytaire mature et, chez les nourrissons et les enfants atteints de sclérose tubéreuse, le rhabdomyome, c'est-à-dire l'hamartome myocardique des ventricules ou du septum interventriculaire. [ 5 ]
L'hamartome cardiomyocytaire mature se développe dans la paroi ventriculaire (et rarement dans les oreillettes) et peut se présenter sous forme de lésions multiples, masses denses étroitement liées au myocarde sous-jacent. La tumeur peut provoquer des symptômes d'insuffisance cardiaque: douleurs thoraciques, palpitations et arythmies, souffles cardiaques, œdème, dyspnée, cyanose.
Les rhabdomyomes cardiaques, dont la plupart sont diagnostiqués au cours de la première année de vie, sont composés de tissu musculaire cardiaque formé de myoblastes embryonnaires et ont l'apparence de masses focales solides sans capsule.
Généralement, ces hamartomes se présentent de manière asymptomatique et régressent spontanément avant l’âge de 4 ans.
Les lésions hamartomateuses sont également considérées par certains experts comme étant associées au myxome du cœur du complexe de Carney. [ 6 ]
Gamartome du tractus gastro-intestinal
L'hamartome gastrique est une masse mésenchymateuse se présentant sous la forme d'un polype épithélial hyperplasique de l'estomac, d'un polype de Peutz-Jeghers ou d'un hamartome myoépithélial rare, caractérisé par une hypertrophie des faisceaux musculaires lisses. On le désigne également sous les noms d'hamartome myoglandulaire, d'hamartome adénomyomateux et d'adénomyome gastrique. Les manifestations cliniques typiques incluent dyspepsie, douleurs épigastriques et saignements gastro-intestinaux hauts. [ 7 ], [ 8 ]
Plus d'informations dans le document - Polypose gastrique
Un hamartome intestinal est un polype hamartomateux ou hyperplasique du gros intestin, diagnostiqué comme un adénome adénomateux ou tubulaire. Lorsque l'hamartome est localisé dans la glande de Brunner du duodénum, les symptômes se manifestent par des douleurs épigastriques; des nausées, des vomissements et des flatulences (signe d'une occlusion intestinale); et, s'il est important, des saignements gastro-intestinaux. En cas d'hamartome myoépithélial de l'iléon, les patients se plaignent de douleurs abdominales, d'une perte de poids et d'une anémie chronique. [ 9 ], [ 10 ]
Lire aussi - Polypes rectaux
L'hamartome rétrorectal est un kyste kystique, ou kyste multichambre de l'espace rétrorectal (tissu conjonctif lâche entre le rectum et son propre fascia), qui survient le plus souvent chez les femmes d'âge moyen. Il se présente sous la forme d'un kyste bombé sur la paroi postérieure du rectum, tapissée d'épithélium et contenant des fibres musculaires lisses disposées de manière chaotique. Cet hamartome se manifeste par des douleurs abdominales basses et une constipation récurrente. [ 11 ], [ 12 ]
Hamartomes du foie et de la rate
L'hamartome biliaire multiple du foie est un hamartome des voies biliaires intrahépatiques interdivisionnaires associé à des malformations de leur développement durant la période embryonnaire. Cet hamartome (unique ou multiple) est constitué d'amas de voies biliaires dilatées de manière anarchique et de stroma fibrocollagène. [ 13 ]
Les hamartomes biliaires sont asymptomatiques et sont généralement découverts fortuitement (lors d'un examen radiologique ou d'une laparotomie). [ 14 ]
L'hamartome de la rate est une tumeur primitive bénigne rare et souvent détectée fortuitement. Il se compose d'éléments de la pulpe rouge de la rate, formant une masse homogène bien définie et de consistance ferme. Cette malformation peut être unique ou multiple; la compression du parenchyme splénique peut provoquer une sensation d'inconfort et de douleur dans la région sous-costale gauche. [ 15 ], [ 16 ]
Hamartomes rénaux
L'hamartome rénal le plus fréquent est l' angiomyolipome rénal, car cette tumeur bénigne est constituée de tissu adipeux mature incrusté de fibres musculaires lisses et de vaisseaux sanguins. Il se forme dans la sclérose tubéreuse de Bourneville dans 40 à 80 % des cas. Une augmentation de la taille de l'hamartome (plus de 4 à 5 cm) entraîne des douleurs et la présence de sang dans les urines. [ 17 ], [ 18 ]
Hamartome du sein
Les définitions diagnostiques de l'hamartome mammaire acceptées par l'OMS sont des termes tels qu'adénolipome, chondrolipome et hamartome myoïde. Bien que souvent appelé fibroadénolipome par les mammologues, il est dû au fait que la tumeur contient des cellules de tissu fibreux, glandulaire et adipeux enfermées dans une fine capsule de tissu conjonctif aux contours distincts. Des calcifications focales peuvent être observées à la visualisation. Dans ce cas, les manifestations cliniques sont absentes. [ 19 ], [ 20 ]
Lire aussi - Tumeurs du sein
Hamartomes du cerveau
Un tiers des patients atteints de sclérose tubéreuse présentent un hamartome cérébral se présentant sous la forme d'excroissances corticales intracrâniennes ou de tubercules dans différents lobes – à la frontière entre la substance grise et la substance blanche – ou de nodules sous-épendymaires le long des parois des ventricules cérébraux. Un hamartome astrocytaire, un astrocytome sous-épendymaire à cellules géantes avec rupture corticale, dysmorphie neuronale et grandes cellules gliales du parenchyme cérébral (astrocytes), peut également se former. Les symptômes des hamartomes cérébraux incluent des crises d'épilepsie et un retard mental chez l'enfant. [ 21 ], [ 22 ]
Une malformation rare qui survient pendant l'embryogenèse et est présente à la naissance est l'hamartome hypothalamique, une masse de neurones hétérotopiques et de cellules gliales. À mesure que le cerveau de l'enfant grandit, la tumeur s'agrandit, mais ne se propage pas à d'autres régions cérébrales. [ 23 ], [ 24 ]
Si des tissus hypertrophiés se forment dans la partie antérieure de l'hypothalamus (tuber cinereum), là où l'hypophyse s'y attache, la malformation se manifeste par des symptômes de développement sexuel prématuré central (avant 8-9 ans): apparition d'éruptions cutanées d'acné, développement précoce des glandes mammaires et ménarche précoce chez les filles; mutation précoce des poils pubiens et de la voix chez les garçons.
Lorsque des hamartomes se forment dans la partie postérieure de l'hypothalamus, il peut y avoir des anomalies dans l'activité électrique du cerveau, qui se manifestent dans la petite enfance par des crises, et à un stade ultérieur (de 4 à 7 ans) par une épilepsie avec des crises d'épilepsie focales avec rire soudain ou avec pleurs involontaires, des crises atoniques et tonico-cloniques, ainsi que des crises d'agression, des troubles de la mémoire et des troubles cognitifs.
Un hamartome hypophysaire est un adénome hypophysaire bénin survenant de façon sporadique.
Les adultes d'âge moyen atteints du syndrome de Cowden peuvent présenter une masse rare ressemblant à une tumeur, un hamartome du cervelet, diagnostiqué comme un gangliocytome cérébelleux dysplasique ou une maladie de Lhermitte-Duclos. Les symptômes peuvent être absents ou se manifester par des céphalées, des vertiges, une altération de la coordination des mouvements et une paralysie de nerfs crâniens.
Hamartome ganglionnaire
Lorsque les cellules du muscle lisse et du tissu adipeux, ainsi que les vaisseaux sanguins et le stroma collagène des ganglions lymphatiques inguinaux, rétropéritonéaux, sous-mandibulaires et cervicaux se développent excessivement, un hamartome angiomyomateux d'un ganglion lymphatique ou un hamartome angiomyomateux nodulaire se forme - avec remplacement partiel ou complet de son parenchyme. [ 25 ], [ 26 ]
Un hamartome de la peau
En présence de sclérose tubéreuse ou de neurofibromatose, divers hamartomes de la peau sont observés, le plus souvent sous forme de taches hypopigmentées; taches de café et de lait; angiofibrome (sur les joues, le menton, les sillons nasogéniens); taches de galuchat de diverses localisations (qui sont des nævus du tissu conjonctif); plaques fibreuses sur le front, le cuir chevelu ou le cou.
Une manifestation dermatologique rare de la sclérose tubéreuse (en particulier chez les hommes) est l'hamartome folliculokystique et collagène, caractérisé par un dépôt abondant de collagène dans le derme, une fibrose périfolliculaire concentrique et des kystes sous-cutanés en forme d'entonnoir remplis de kératine observés à l'examen histopathologique. [ 27 ]
Aux hamartomes constitués de mélanocytes (cellules qui produisent le pigment mélanine), la plupart des experts font également référence à divers néoplasmes mélanocytaires, en particulier les nævus mélanocytaires congénitaux, qui représentent une anomalie de l'embryogenèse.
En termes d'étiologie, les hamartomes composés de tissu vasculaire sont également des hémangiomes de la peau.
Les patients atteints du syndrome de Peutz-Jeghers-Thuren présentent un hamartome sous forme de pigmentation inégale de la peau et des muqueuses - lentiginose périorificielle
Les cas d'hamartome ectodermique-mésodermique papuleux linéaire (Hamartoma moniliformis) présentent une éruption papuleuse linéaire de couleur chair sur la tête, le cou et la partie supérieure de la poitrine.
Et un hamartome sébocytaire est un hamartome des glandes sébacées, en savoir plus dans la publication - naevus sébacé.
Hamartome de l'œil
Les lésions hamartomateuses pigmentées de l'iris dans la neurofibromatose de type 1 et le syndrome de Watson – se présentant sous la forme d'amas nodulaires de mélanocytes dendritiques – sont appelées hamartomes iriens ou nodules de Lisch. Il s'agit de papules jaune-brun arrondies, transparentes (généralement sans incidence sur la vision), en forme de dôme, qui font saillie à la surface de l'iris.
Les patients atteints d'angiofibrome juvénile du nasopharynx et de polypose adénomateuse familiale développent souvent un hamartome combiné de la rétine et de l'épithélium pigmentaire rétinien - sous la forme d'une tache noire sur la partie centrale (maculaire) de la rétine. [ 28 ]
Un hamartome du nez
L'hamartome nasal est défini par les spécialistes comme un hamartome chondro-mésenchymateux nasal, ou chondrome nasal, dû à une prolifération bénigne de l'épithélium respiratoire, des glandes sous-muqueuses et du mésenchyme chondro-osseux. Ses manifestations cliniques dépendent de la taille et de la localisation de la lésion et comprennent: congestion nasale, difficultés respiratoires et d'allaitement chez le nourrisson, écoulement nasal clair et aqueux et saignements de nez. Un hamartome peut grandir avec l'enfant et se propager aux orbites, entraînant un déplacement du globe oculaire vers l'avant ou vers l'arrière, un strabisme ou des troubles oculomoteurs. [ 29 ]
Un hamartome chez un enfant
Toutes les lésions hamartomateuses mentionnées ci-dessus de divers organes et structures anatomiques sont présentes chez les enfants présentant des syndromes correspondants.
Les nouveau-nés présentent un hamartome mésenchymateux de la paroi thoracique ou un hamartome cartilagineux des côtes. Il s'agit de masses solides et immobiles résultant d'une prolifération focale d'éléments squelettiques normaux par des éléments cartilagineux, vasculaires et mésenchymateux. Cet hamartome peut entraîner une insuffisance respiratoire et le développement d'un syndrome de détresse respiratoire. L'hamartome mésenchymateux du foie est la deuxième tumeur hépatique bénigne la plus fréquente chez l'enfant. Cette formation pseudo-tumorale (le plus souvent localisée dans le lobe droit de l'organe) est constituée de cellules du stroma mésenchymateux, d'hépatocytes et de cellules épithéliales de la paroi des voies biliaires. Le tableau clinique comprend une masse palpable dans la cavité abdominale, une anorexie et une perte de poids. En cas de taille importante (jusqu'à 10 cm et plus), la tumeur recouvre les voies biliaires extra-hépatiques et la veine cave inférieure, ce qui entraîne un ictère et un œdème des membres inférieurs.
Un hamartome est un néphrome mésoblastique congénital (survenant chez 1 nourrisson sur 200 000) qui peut entraîner un ballonnement abdominal chez le nouveau-né, accompagné d'une masse palpable de consistance dense dans le quadrant supérieur droit de l'abdomen. Les nourrissons peuvent également présenter une respiration rapide et superficielle.
Les anomalies congénitales rares comprennent l'hamartome fibreux de la petite enfance, qui survient chez les enfants au cours des deux premières années de vie et se présente comme une masse nodulaire indolore dans les tissus sous-cutanés de l'aisselle, du cou, de l'épaule et de l'avant-bras, du dos et de la poitrine, de la cuisse, du pied et des organes génitaux externes.
L'hamartome angiomateux eccrine chez l'enfant peut être présent dès la naissance ou se manifester dès la petite enfance. Cette tumeur bénigne de nature hamartomateuse se présente généralement sous forme de nodules et/ou de plaques bleuâtres ou brunâtres, résultant de la prolifération des glandes sudoripares eccrines et des capillaires dans les couches moyennes et profondes du derme. Cet hamartome peut provoquer une hyperhidrose localisée et une pilosité accrue.
Complications et conséquences
Il est généralement admis que les hamartomes récidivent rarement et se transforment rarement en tumeurs malignes. Ils présentent souvent peu ou pas de symptômes et disparaissent parfois avec le temps. Cependant, dans les cas plus graves et selon leur localisation, ces malformations peuvent entraîner de graves complications et conséquences.
Tout d’abord, un hamartome peut atteindre une taille telle qu’il commence à exercer une pression sur les tissus et les organes environnants, perturbant ainsi leurs fonctions.
L'hamartome cardiaque chez les enfants peut entraîner des anomalies persistantes du rythme cardiaque, des anomalies valvulaires et une altération du flux sanguin intracardiaque avec insuffisance cardiaque congestive ultérieure.
Les complications des polypes hamartomateux du tube digestif sont des saignements gastro-intestinaux, une occlusion et une invagination intestinale (avec une possible issue fatale). Un hamartome rénal volumineux peut provoquer une rupture rénale.
Un hamartome dans le cerveau peut provoquer un syndrome d’hydrocéphalie obstructive.
Dans les hamartomes hypothalamiques et hypophysaires, la production d'hormone somatotrope (hormone de croissance) peut être altérée, entraînant le développement d' un nanisme hypophysaire (hypopituitarisme) chez l'enfant. Les hamartomes hypothalamiques chez l'enfant peuvent également entraîner une épilepsie pharmacorésistante.
Les complications de l'hamartome de l'épithélium pigmentaire rétinien sont lourdes de dysfonctionnement de la rétine et/ou du nerf optique, d'œdème maculaire, de néovascularisation de la choroïde et de décollement de la rétine.
Diagnostics ng hamartomas
Une partie importante du diagnostic des hamartomes et des syndromes apparentés est la collecte de l'anamnèse, y compris les antécédents familiaux.
Les examens de laboratoire comprennent des analyses sanguines: examen clinique général; électrolytes sériques; profil lymphocytaire; taux de calcium, de potassium, de phosphate et d’urée; et tests de la fonction hépatique. Si possible, une ponction-biopsie de la masse est réalisée, l’examen histologique étant crucial pour le diagnostic et le choix du traitement.
Le diagnostic instrumental permet de visualiser la formation de type tumoral hamartomateux et d'identifier sa localisation exacte, pour laquelle on utilise la radiographie, l'angiographie, l'électroencéphalographie (EEG), l'échographie (sonographie), la tomodensitométrie (TDM), la TEP (tomographie par émission de positons), l'IRM (imagerie par résonance magnétique).
Diagnostic différentiel
Devant toute masse anormale, le diagnostic différentiel est essentiel. Ainsi, on distingue le tuberculome de l'hamartome; l'hamartome pulmonaire du cancer du poumon primitif, du carcinoïde bronchique et des métastases. L'hamartome cérébral doit être distingué du craniopharyngiome et du gliome hypothalamo-chiasmatique. Le diagnostic différentiel de l'hamartome avec le néphrome mésoblastique congénital inclut la tumeur de Wilms (néphroblastome malin), le sarcome à cellules claires du rein et la tumeur rénale ossifiante du nourrisson.
Qui contacter?
Traitement ng hamartomas
Si l'hamartome est asymptomatique et découvert fortuitement, aucun traitement n'est nécessaire, mais il est nécessaire de surveiller son évolution et l'état du patient. Dans d'autres cas, le traitement vise à réduire l'intensité des symptômes et à prévenir les complications. Par exemple, en cas d'hamartome hypothalamique présentant des symptômes de puberté précoce, des médicaments inhibant la libération de certaines hormones sont prescrits. Les médicaments cardiaques sont utilisés pour traiter les symptômes d'insuffisance cardiaque chez les patients atteints d'hamartomes cardiaques.
L'ablation chirurgicale des hamartomes est indiquée pour confirmer le diagnostic et en cas de symptômes intenses médicalement non corrigibles.
Par exemple, les hamartomes pulmonaires peuvent être réséqués par résection cunéiforme et, dans les cas graves, par l'ablation d'un lobe pulmonaire (lobectomie). Un hamartome mammaire peut également être excisé et, s'il est volumineux, une mastectomie partielle ou complète peut être nécessaire.
La thermoablation par radiofréquence stéréotaxique ou l'ablation laser peuvent être utilisées pour retirer les polypes hamartomateux. La radiochirurgie par rayons gamma hautement focalisés (gamma knife pour les hamartomes hypothalamiques ou astrocytaires) est également utilisée.
La prévention
La seule méthode de prévention du développement des hamartomes peut être considérée comme un dépistage génétique des futurs parents de l'enfant.
Prévoir
Le pronostic global de cette anomalie congénitale dépend de la localisation et de la taille du néoplasme, ainsi que des comorbidités et de l’état de santé général du patient.