Expert médical de l'article

Nouvelles publications
Osteoblastoblastoma
Dernière revue: 29.06.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
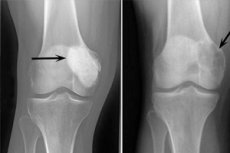
L'ostéoblastoclastome est un processus tumoral bénin ou malin qui endommage différents os du squelette. Initialement appelée tumeur gigantocellulaire (depuis 1912), cette pathologie fut nommée dix ans plus tard « ostéoclastome ». Ce n'est qu'en 1924 que le professeur Rusakov introduisit le terme affiné « ostéoblastoclastome », plus adapté à la composition cellulaire de la tumeur.
Aujourd'hui, l'ostéoblastoclastome est considéré comme une véritable tumeur des tissus mous dotée d'un réseau vasculaire étendu. La seule option thérapeutique appropriée est l'ablation de la tumeur au sein des tissus sains, parfois associée à une greffe osseuse. [ 1 ]
Épidémiologie
L'incidence des tumeurs osseuses dans le monde varie de 0,5 à 2 %. Selon les statistiques américaines, l'ostéosarcome (environ 34 % des cas), le chondrosarcome (27 %) et la tumeur d'Ewing (18-19 %) sont les plus fréquents. Les chordomes, les fibrosarcomes, les histiocytomes, les tumeurs à cellules géantes et les angiosarcomes sont moins fréquents.
Le taux d'incidence est fortement corrélé à l'âge. Ainsi, la première poussée de croissance tumorale est détectée à l'adolescence (vers 16 ans), et la seconde à l'âge mûr.
L'ostéoblastoclastome est une tumeur relativement fréquente. Il survient dans environ 2 à 30 % des néoplasies osseuses. Les femmes sont plus souvent touchées, mais les hommes peuvent également l'être, principalement entre 18 et 40 ans. Les enfants de moins de 12 ans sont rarement touchés, mais même à cet âge, l'incidence n'est pas exclue. Des cas familiaux et héréditaires d'ostéoblastoclastome ont été décrits.
Le plus souvent (environ 75 %), la tumeur se trouve dans les os tubulaires longs, beaucoup moins souvent les os plats et petits sont touchés.
Dans les os longs tubulaires, l'épimétaphyse est principalement touchée, et chez l'enfant, c'est la métaphyse qui est touchée. La tumeur ne se développe pas dans la zone des cartilages épiphysaires et articulaires. Très rarement, le problème touche la diaphyse (moins de 0,5 % des cas).
On constate qu'avec les progrès de la médecine, l'incidence de l'ostéoblastoclastome reste stable, mais les taux de mortalité ont considérablement diminué. La cause principale et la plus probable de cette pathologie est considérée comme l'impact des rayonnements ionisants. Ainsi, les risques sont accrus chez les personnes ayant reçu de fortes doses de radiothérapie, ainsi que chez celles ayant reçu des injections de radio-isotopes (à des fins diagnostiques ou thérapeutiques). Parmi les autres facteurs étiologiques fréquents figurent une écologie défavorable et l'hérédité. [ 2 ]
Causes ng osteoblastoblastomas
L'ostéoblastoclastome est un foyer de cellules pathologiquement altérées pouvant apparaître dans presque toutes les parties du squelette. Malgré les anomalies structurelles, les cellules pathologiques continuent de se diviser, comme dans les tissus sains. Leur structure diffère largement de la normale, ce qui entraîne le remplacement des propriétés de l'os directement affecté et de sa fonction typique. Les cellules malignes pathologiquement altérées acquièrent une propension à une multiplication incontrôlée, souvent rapide, entraînant une augmentation du volume tumoral. Le tissu osseux auparavant normal peut être déplacé par les structures du néoplasme, et des cellules pathologiques individuelles peuvent être séparées et transportées par le sang ou la lymphe vers d'autres zones anatomiques distantes. C'est ainsi que des métastases se forment.
Il est connu que l'ostéoblastoclastome malin peut provenir de n'importe quelle tumeur maligne localisée dans le corps (y compris les tumeurs des organes internes). Le processus se propage par métastase. Cependant, la plupart des ostéoblastoclastomes (bénins et malins) sont des néoplasies primaires qui apparaissent et se développent en premier lieu au même endroit.
En général, les ostéoblastoclastomes sont des tumeurs multifactorielles dont les causes exactes n'ont pas encore été établies. Les conditions favorisant l'apparition d'une néoplasie sont notamment:
- Un état d’immunodéficience;
- Modifications tissulaires congénitales;
- Influences environnementales mutagènes;
- Changements hormonaux;
- Pathologies et blessures concomitantes (un traumatisme est souvent présent dans l'anamnèse).
Facteurs de risque
On manque de données précises sur les causes de la formation d'ostéoblastoclastomes. Cependant, les experts suggèrent l'implication de plusieurs facteurs associés à un risque accru d'oncopathologies osseuses:
- Hérédité. Dans de nombreux cas, la prédisposition aux processus tumoraux est génétiquement déterminée. Cela pourrait notamment être le cas du syndrome de Leigh-Fraumeni, qui prédispose au développement de diverses tumeurs, notamment cancéreuses et sarcomes.
- Maladie de Paget. Cette maladie peut affecter un ou plusieurs os et fait partie des pathologies prétumorales. Chez les patients atteints de cette affection, les os s'épaississent et deviennent simultanément fragiles, ce qui entraîne de fréquentes fractures pathologiques. Les ostéosarcomes surviennent dans environ 8 % des cas de maladie de Paget sévère.
- Excroissances osseuses multiples, exostoses.
- Ostéochondromes multiples (y compris héréditaires).
- Enchondromes multiples (risque faible mais toujours présent).
- Exposition aux radiations (y compris les radiations intenses utilisées pour traiter d’autres processus tumoraux et les effets du radium et du strontium radioactifs).
Une catégorie particulière de risque comprend la radiothérapie pendant l’enfance et le jeune âge, recevant des doses supérieures à 60 Gray.
Les experts attirent l'attention sur le fait que les rayons non ionisants - en particulier les micro-ondes et les rayonnements électromagnétiques, qui se forment à partir des lignes électriques, des téléphones portables et des appareils électroménagers - ne comportent pas de risques d'ostéoblastoclastome.
Pathogénèse
Les caractéristiques pathogéniques de l'apparition et du développement de l'ostéoblastoclastome ne sont pas entièrement comprises, en raison de la complexité de la pathologie. La cause principale de la formation tumorale est un défaut de différenciation cellulaire dû à un dysfonctionnement du système immunitaire. Ceci entraîne la croissance d'une tumeur composée de cellules indifférenciées, qui déterminent les propriétés de la tumeur et ressemblent structurellement à des cellules immatures. Si la structure cellulaire est proche de la normale, mais ne l'est pas, on parle d'ostéoblastoclastome bénin. En cas de modifications importantes de la structure cellulaire, la tumeur est attribuée à des processus malins. Une telle tumeur se caractérise par une modification du repliement antigénique des cellules, une croissance et une division cellulaires incontrôlées. Outre la perte de spécificité de la structure cellulaire, la fonctionnalité est également affectée. L'ostéoblastoclastome malin se distingue notamment de l'ostéoblastoclastome bénin par son invasion des tissus sains voisins. Dans le cas d'une tumeur osseuse bénigne, il n'y a pas de germination dans des structures saines, il n'y a pas de tendance à une croissance rapide et à une propagation dans tout le corps, il n'y a pas de tendance à l'autodestruction arbitraire et à l'intoxication par les produits de décomposition tumorale.
Dans tous les cas, la structure osseuse est détruite, quelle que soit la bénignité de la pathologie. Le segment osseux affecté devient alors fragile et cassant. Une fracture pathologique, même sous une charge minimale, est souvent à l'origine de la consultation médicale.
Il est important de noter: la bénignité du processus est toujours un état conditionnel, car il existe des risques de malignité, et le foyer bénin se transforme, un ostéoblastoclastome malin se produit.
Symptômes ng osteoblastoblastomas
Le tableau clinique de l'ostéoblastoclastome dépend principalement de la localisation et du stade du processus pathologique. En règle générale, la tumeur présente les caractéristiques suivantes:
- Le néoplasme est solitaire;
- Affecte principalement les os tubulaires des membres inférieurs ou supérieurs;
- On le trouve moins fréquemment dans les os plats;
- Il y a une douleur lancinante dans le segment affecté;
- Le motif cutané et vasculaire au-dessus du foyer pathologique augmente;
- Le membre malade est déformé (augmentation de volume localisée);
- Le travail de l’articulation la plus proche de l’ostéoblastoclastome ou du membre dans son ensemble est perturbé;
- Foyer compact déterminé par palpation avec un « craquement de parchemin » caractéristique.
En général, les symptômes peuvent être classés en symptômes locaux et généraux. Les symptômes locaux sont détectés visuellement, notamment par la présence d'une courbure ou d'un bombement du fragment osseux. L'attention est également attirée sur l'altération cutanée au-dessus du foyer pathologique: un schéma vasculaire est clairement visible, les tissus sont gonflés ou aplatis. La tumeur peut être palpée; elle est souvent indolore, mais présente une structure caractéristique. Les tumeurs malignes sont généralement nodulaires et de configuration irrégulière.
L'articulation adjacente peut être limitée dans ses mouvements et souffrir de douleurs persistantes. La compression des vaisseaux et des troncs nerveux entraîne souvent une altération de la sensibilité et un gonflement persistant. Le système lymphatique réagit également: les ganglions lymphatiques voisins se dilatent.
La symptomatologie générale est plus typique des ostéoblastoclastomes malins et est due aux processus d'intoxication de l'organisme. Les patients peuvent présenter:
- Fièvre, états fébriles;
- Maigreur;
- Faiblesse constante;
- Somnolence ou insomnie, troubles de l’appétit;
- Transpiration excessive nocturne;
- Effondrement.
Il existe également un faible pourcentage d'ostéoblastoclastomes, généralement de petite taille et non apparents cliniquement. Leur découverte est fortuite lors d'examens radiologiques ou d'imagerie pour d'autres raisons.
Premiers signes d'ossification d'ostéoblastoclastome
- Accélération de la croissance du néoplasme.
- Syndrome de douleur accrue.
- Expansion du foyer destructeur en diamètre, ou transformation de la forme cellulaire-trabéculaire en forme lytique.
- Désintégration de la couche corticale sur une zone relativement longue.
- Perte de clarté des configurations du foyer destructeur.
- Désintégration de la plaque de fermeture qui bloquait le canal médullaire.
- Réaction périostée.
La malignité de l'ostéoblastoclastome est basée sur des indicateurs cliniques et radiologiques et est nécessairement confirmée par un diagnostic morphologique des tissus tumoraux.
Outre l'osloplastie d'une tumeur initialement bénigne, on observe également un ostéoblastoclastome malin primitif. Il s'agit en fait d'un sarcome d'étiologie ostéogénique.
La localisation de l'ostéoblastoclastome malin est identique à celle d'un ostéoblastoclastome bénin. La radiographie révèle un foyer destructeur dans le tissu osseux, sans contours nets. La destruction de la couche corticale est étendue, s'étendant souvent aux tissus mous.
Signes permettant de distinguer l’ostéoblastoclastome malin de la forme ostéogénique du sarcome ostéoclastique:
- L’âge majoritairement avancé des patients;
- Symptomatologie moins vive;
- Un pronostic à long terme plus favorable.
Ostéoblastoclastome chez les enfants
L'ostéoblastoclastome chez l'enfant est rare: on ne recense que deux ou trois cas pour un million d'enfants. Il convient de noter que parmi tous les patients pédiatriques, ce sont les plus de 10-15 ans qui prédominent.
Les scientifiques ne parviennent pas à identifier la cause exacte de l'ostéoblastoclastome chez l'enfant. Cette pathologie est vraisemblablement liée à une croissance rapide de l'enfant, ainsi qu'à un facteur génétique.
Il existe également des indices de causes possibles telles que l'exposition aux radiations (notamment la radiothérapie) et la chimiothérapie (prise de cytostatiques). De nombreux médicaments de chimiothérapie peuvent détruire le matériel génétique des cellules osseuses, ce qui conduit au développement d'une tumorigenèse.
De plus, le risque d'ostéoblastoclastome est plus élevé chez les enfants atteints de certaines maladies congénitales, comme le rétinoblastome bilatéral ou le syndrome de Li-Fraumeni. Un lien de causalité existe également avec la maladie de Paget.
On sait également que chez la grande majorité des enfants (environ 90 %), les médecins ne parviennent à détecter aucun des facteurs de risque mentionnés ci-dessus.
Il est difficile de prédire l'évolution de l'ostéoblastoclastome dans l'enfance, car elle dépend des caractéristiques d'une tumeur particulière, de sa localisation, du degré de propagation au moment du diagnostic, de la rapidité du traitement et de l'exhaustivité de l'élimination du néoplasme.
La qualité du traitement de l'ostéoblastoclastome a considérablement progressé au cours des deux ou trois dernières décennies. Le protocole thérapeutique est désormais combiné et le taux de guérison a dépassé 70 à 80 %. Une issue favorable est obtenue si la tumeur est retirée chirurgicalement et que son effet est consolidé par une chimiothérapie suffisante. Les enfants atteints d'ostéoblastoclastome bénin ont les meilleures chances de guérison.
Lorsque l'on annonce des chiffres précis sur la guérison, on ne s'en tient qu'à des chiffres généraux: aucune statistique ne permet de prédire et de déterminer avec précision les chances de guérison d'un enfant en particulier. Le terme « guérison » désigne principalement « l'absence de processus tumoral dans l'organisme », les approches thérapeutiques modernes garantissant une absence de récidive à long terme. Cependant, il ne faut pas oublier le risque d'effets secondaires indésirables et de complications tardives. Par conséquent, tout traitement, quelle que soit sa complexité, doit s'accompagner de mesures de rééducation de haute qualité. De plus, les enfants ont besoin de soins orthopédiques pendant une longue période.
Formes
La classification des néoplasies osseuses est assez large. L'attention est principalement portée sur les variations de structure cellulaire et les caractéristiques morphologiques du processus tumoral. Ainsi, les tumeurs sont divisées en deux catégories:
- Ostéogénique (formé à partir de cellules osseuses);
- Néostéogène (formé dans l'os sous l'influence d'autres types de cellules - par exemple, des structures vasculaires ou du tissu conjonctif).
L'ostéoblastoclastome osseux est une tumeur essentiellement bénigne. Cependant, malgré cela, sa croissance est souvent agressive et contribue à la destruction et à l'amincissement des tissus osseux, ce qui rend une intervention chirurgicale obligatoire. L'ostéoblastoclastome à cellules géantes peut également être malin.
En fonction des paramètres cliniques et radiologiques et du tableau morphologique, on distingue trois formes fondamentales d'ostéoblastoclastomes:
- La forme cellulaire se rencontre principalement chez les personnes âgées et se caractérise par une évolution lente. Le diagnostic révèle une tuméfaction épaissie et grumeleuse, sans possibilité de délimitation clinique du foyer tumoral par rapport aux zones osseuses saines.
- La forme kystique se manifeste d'abord par une douleur. À la palpation, on observe un « crissement de parchemin ». À l'examen visuel, on observe une tumeur osseuse de forme convexe et bombée.
- La forme lytique est considérée comme une variante rare de la pathologie; elle est principalement détectée à l'adolescence. Le processus tumoral se développe assez rapidement et le patient commence à ressentir des douleurs, notamment à la palpation.
Une tumeur à cellules géantes peut se former sur presque tous les os du squelette, bien que les os tubulaires des membres, des côtes et de la colonne vertébrale soient un peu plus souvent touchés. L'ostéoblastoclastome de la mâchoire inférieure est deux fois plus fréquent que celui de la mâchoire supérieure. À la palpation, on observe une tumeur dense avec des zones ramollies. Les patients se plaignent le plus souvent de la présence d'une protubérance qui saigne et crée une gêne lors de la mastication. À mesure que le problème progresse, il s'accompagne d'une altération de la fonction de l'articulation temporo-mandibulaire. Parmi les os tubulaires, la tumeur affecte plus souvent le fémur et le tibia. L'ostéoblastoclastome du fémur survient principalement chez les personnes d'âge moyen. La maladie s'accompagne d'une altération de la fonction de l'articulation correspondante, d'une boiterie et d'une vascularisation cutanée prononcée.
Outre la classification ci-dessus, il existe des formes centrales et périphériques de pathologie, bien qu'il n'existe aucune différence morphologique entre elles. L'ostéoblastoclastome périphérique a une localisation gingivale, tandis que la forme centrale se développe dans l'os et se distingue par la présence d'hémorragies multiples (d'où le surnom de tumeur brune de l'ostéoblastoclastome central). L'apparition d'une couleur brune est due au dépôt d'érythrocytes, qui se désintègrent pour former de l'hémosidérine.
Les néoplasmes osseux malins passent par les étapes suivantes dans leur développement:
- Un foyer T1 mesurant 3 à 5 cm est situé dans l'os et un segment musculofascial.
- Les foyers T2 ne s'étendent pas sur plus de 10 cm le long de l'os mais ne s'étendent pas au-delà d'un cas fascial.
- Les foyers T3 quittent les limites d'un cas musculo-fascial et se propagent dans un autre cas voisin.
- Les foyers T4 naissent de la peau ou des troncs neurovasculaires.
De la même manière, le degré d’atteinte des ganglions lymphatiques et la propagation des métastases sont catégorisés.
Complications et conséquences
Parmi les complications de l'ostéoblastoclastome figure l'augmentation de l'activité du néoplasme, qui survient souvent après une longue période de silence. Dans certains cas, on parle de dégénérescence maligne du processus tumoral ou de sa propagation dans des structures anatomiques proches et sensibles.
- La propagation au tronc nerveux provoque l'apparition d'un syndrome douloureux neuropathique en raison de son effet sur le nerf de gros calibre. Cette douleur persiste pratiquement après la prise d'analgésiques conventionnels, ce qui épuise littéralement le patient.
- La propagation aux vaisseaux sanguins peut être compliquée par un saignement massif et soudain et la formation d’un hématome.
Des complications ne sont pas exclues, qui s'accompagnent d'une violation de la fonction des articulations voisines: la croissance de l'ostéoblastoclastome dans une telle situation bloque le fonctionnement adéquat du mécanisme musculo-squelettique, ce qui conduit à une amplitude de mouvement limitée et à l'apparition d'un syndrome douloureux.
Les complications les plus fréquentes de l'ostéoblastoclastome sont des fractures pathologiques de la zone affectée. Ce problème survient même après un traumatisme mineur, car le tissu osseux devient extrêmement fragile et instable.
De plus, les spécialistes évoquent également des effets indésirables généraux et locaux spécifiques caractéristiques de l'ostéoblastoclastome malin:
- La formation de métastases distantes et proches;
- Intoxication du corps par des produits de décomposition.
Si des métastases sont détectées quelque temps après les mesures diagnostiques initiales, cela indique l’inefficacité du traitement en cours et la progression du néoplasme.
Une autre ligne de complications est l'apparition d'une nouvelle tumeur ou d'une pathologie générale due à la chimiothérapie ou à l'irradiation du foyer osseux d'ostéoblastoclastome.
Diagnostics ng osteoblastoblastomas
Les méthodes de diagnostic utilisées pour détecter l'ostéoblastoclastome comprennent:
- Clinique, qui comprend l’examen externe et la palpation de la zone pathologiquement altérée;
- Radiographie (radiographie antéropostérieure et latérale, si indiquée - radiographie ciblée et oblique);
- Tomographique (utilisant l'imagerie par résonance magnétique ou informatisée);
- Radio-isotope;
- Morphologique, qui comprend l'analyse histologique, histochimique et cytologique du biomatériau obtenu lors d'une ponction ou d'une trépanobiopsie;
- Laboratoire.
Le médecin étudie attentivement l'histoire de la maladie, identifie les premiers signes, précise la localisation et le type de syndrome douloureux, ses caractéristiques, prend en compte les résultats des examens et traitements antérieurs et évalue l'état général du patient. En cas de suspicion de pathologie des os longs tubulaires, le spécialiste prête attention à la présence d'un œdème, d'une limitation motrice des articulations les plus étroites, ainsi qu'à la présence de symptômes neurologiques, d'une faiblesse et d'une hypotrophie musculaires. Il est important d'examiner attentivement les organes internes afin de détecter une éventuelle propagation de métastases.
Tous les patients subissent des analyses sanguines et urinaires générales, avec dosage des protéines et des fractions protéiques, du phosphore, du calcium et des acides sialiques. Il est également nécessaire de déterminer l'activité enzymatique des phosphatases, de réaliser un test de Definil et d'étudier l'indice de protéine C-réactive. Pour différencier un ostéoblastoclastome d'un myélome, le patient subit une analyse urinaire pour rechercher la présence de la protéine de Bence-Jones pathologique.
Le diagnostic radiologique est fondamental pour le diagnostic de l'ostéoblastoclastome. Un examen radiologique ciblé et une tomographie de haute qualité, obligatoires, permettent de préciser la localisation, le type de foyer pathologique et son extension à d'autres tissus et organes. Grâce à la tomodensitométrie, il est possible de préciser l'état des tissus mous et des structures osseuses les plus fines dans le plan requis, d'identifier les foyers profonds de destruction pathologique, de décrire leurs paramètres dans les limites osseuses et de déterminer le degré de lésion des tissus environnants.
Parallèlement, l'IRM est considérée comme la procédure diagnostique la plus informative, présentant de nombreux avantages par rapport à la radiographie et à la tomodensitométrie. Cette méthode permet d'examiner même les couches tissulaires les plus fines et de dresser un tableau des lésions pathologiques grâce à une image tridimensionnelle spatiale.
Le diagnostic instrumental obligatoire est représenté par des études morphologiques. Le biomatériau obtenu par aspiration et trépanobiopsie, ou par résection de segments osseux avec la tumeur, est évalué. Une biopsie par ponction est réalisée à l'aide d'aiguilles spéciales et sous contrôle radiologique.
Les signes radiologiques suivants sont considérés comme typiques de l’ostéoblastoclastome:
- Limitation de la porosité;
- Homogénéité de la lyse osseuse dans le type de trabéculisation fine;
- La présence de lueurs pseudokystiques qui ont la structure de « bulles de savon » particulières.
Ce tableau radiologique s'accompagne d'une absence de périostose ostéoformative réactive primaire ou secondaire. On observe un amincissement et une atrophie de la couche corticale.
L'ostéoblastoclastome malin, résultant d'une vascularisation intense, entraîne une augmentation de la stase veineuse. Les modifications vasculaires ressemblent à un néoplasme à forte vascularisation.
Diagnostic différentiel
Il est parfois très difficile d'identifier un ostéoblastoclastome. Des problèmes surviennent lors du diagnostic différentiel entre un sarcome ostéogénique et des kystes osseux chez des patients d'âges différents. Selon les statistiques, dans plus de 3 % des cas, l'ostéoblastoclastome a été confondu avec un sarcome ostéogénique et dans près de 14 % des cas avec un kyste osseux.
Le tableau résume les principaux signes de ces pathologies:
Indicateurs |
Ostéoblastoclastome |
Sarcome ostéoplasique ostéogénique |
Kyste osseux |
Âge d'incidence le plus courant |
20 à 30 ans |
20 à 26 ans |
Enfants de moins de 14 ans |
Emplacement |
Région épimétaphysaire |
Région épimétaphysaire |
Zone de métadiaphyse |
Reconfiguration osseuse |
Renflement asymétrique sévère. |
Petite expansion transversale |
Un renflement en forme de fuseau. |
Configuration du foyer destructeur |
Les contours sont clairs |
Les contours sont flous, il n'y a pas de clarté |
Les contours sont clairs |
L'état du canal rachidien |
Couvert par une plaque de fermeture |
Ouvert à la frontière avec le néoplasme |
Aucun changement. |
État de la couche corticale |
Mince, fibreux, discontinu. |
Éclaircie, ruinée |
Mince, plat |
Phénomènes de sclérose |
Atypique |
Présent |
Atypique |
Réaction périostée |
Absent |
Présent sous forme de « visière périostée » |
Absent |
L'état de l'épiphyse |
Le limbe est fin et ondulé. |
Au stade initial, une partie de l'épiphyse reste intacte |
Aucun changement. |
Coupe osseuse à proximité |
Aucun changement. |
Signes d'ostéoporose |
Aucun changement. |
Une attention obligatoire nécessite des indicateurs tels que l'âge du patient, la durée de la pathologie, la localisation du foyer affecté, d'autres informations anamnestiques indiquées dans le tableau.
Les erreurs de diagnostic suivantes sont les plus courantes lorsque l’ostéoblastoclastome est confondu avec de tels processus pathologiques:
- Kyste anévrismal (localisé dans la diaphyse ou la métaphyse des os tubulaires longs);
- Ostéodysplasie fibreuse de type monoaxial (se manifestant principalement dans l’enfance, accompagnée d’une courbure osseuse sans ballonnement osseux);
- Ostéodystrophie hyperparathyroïdienne (pas de délimitation claire du foyer par rapport à la zone osseuse saine, pas de renflement osseux net);
- Métastase osseuse cancéreuse solitaire (caractérisée par des foyers destructeurs aux contours courbes « mangés »).
Il est important de garder à l'esprit que l'ostéoblastoclastome bénin peut toujours se transformer et devenir malin. Les causes de la malignité n'ont pas encore été précisément déterminées, mais les scientifiques pensent que les traumatismes et les changements hormonaux (par exemple, pendant la grossesse) y contribuent. Selon certaines observations, une malignité a également été observée lors de séries répétées de radiothérapies à distance.
Symptômes de l'ossification:
- Le néoplasme commence à se développer rapidement;
- La douleur s'aggrave;
- La taille du foyer destructeur augmente et la phase cellulaire-trabéculaire passe à la phase lytique;
- La couche corticale est en train de se dégrader;
- Les contours du foyer destructeur deviennent indistincts;
- La plaque de verrouillage s'effondre;
- Il y a une réaction périostée.
Lors de la différenciation entre une tumeur maligne primitive (sarcome ostéoclastique ostéogénique) et un ostéoblastoclastome malin, une attention particulière est portée à la durée de l'évolution pathologique et à l'évaluation du tableau radiologique en dynamique. Sur la radiographie d'une tumeur maligne primitive, on ne constate pas de protrusion osseuse typique d'un ostéoblastoclastome, ni de ponts osseux, et on peut observer une zone sclérosée aux contours flous. En cas de malignité, cependant, on observe souvent une petite zone de la plaque de fermeture, qui servait auparavant de barrière au segment osseux sain.
Qui contacter?
Traitement ng osteoblastoblastomas
Le seul traitement approprié pour les patients atteints d'ostéoblastoclastome est la chirurgie. L'intervention la plus douce, pratiquée aux premiers stades du développement tumoral, consiste en l'excision des tissus affectés et le remplissage de la cavité par un greffon. Le greffon est prélevé sur un autre os sain du patient. Cette intervention est la plus favorable et la moins traumatisante, mais elle est aussi, dans certains cas, moins radicale. L'excision du fragment osseux affecté et de la tumeur est considérée comme une méthode plus fiable, réduisant au minimum le risque de récidive tumorale.
S'il s'agit d'un ostéoblastoclastome négligé de grande taille, particulièrement sujet à la malignité ou déjà malin, une amputation partielle ou complète du membre est souvent envisagée.
En général, la tactique du traitement chirurgical de l'ostéoblastoclastome est choisie en fonction de la localisation, de la propagation et de l'agressivité du foyer pathologique.
Si la tumeur affecte les os tubulaires longs, il est alors recommandé de prêter attention à ces types d'interventions chirurgicales:
- Résection des bords avec alloplastie ou autoplastie pour les processus bénins, retardés, les foyers à structure cellulaire et situés en périphérie de l'épimétaphyse. Fixation par vis métalliques.
- Lorsque l'ostéoblastoclastome cellulaire s'étend jusqu'au milieu du diamètre osseux, les deux tiers du condyle, une partie de la diaphyse et la surface articulaire sont réséqués. Le défaut est comblé par une allogreffe de cartilage articulaire. La fixation est assurée par des boulons et des vis.
- En cas de dégradation de l'épimétaphyse sur toute sa longueur ou de fracture pathologique, on utilise des techniques telles que la résection segmentaire avec excision articulaire et le comblement du défaut par une allogreffe. La fixation est assurée par une tige cimentée.
- En cas de fracture pathologique et de malignisation d'un ostéoblastoclastome dans la région proximale du fémur, une arthroplastie totale de la hanche est réalisée.
- En cas de résection des extrémités au niveau de la zone articulaire du genou, la technique de transplantation d'allopolysubstance avec fixation est utilisée. Une endoprothèse totale avec tige allongée en titane est souvent privilégiée pour assurer une radiothérapie ultérieure.
- Si le foyer pathologique est situé à l'extrémité distale du tibia, une résection avec arthrodèse ostéoplastique de la cheville est réalisée. Si le talus est atteint, il est extirpé par arthrodèse en extension.
- En cas de lésions du rachis cervical, un accès antérieur aux vertèbres C1 et C2 est réalisé. Un accès antérolatéral est privilégié. Au niveau Th1-Th2, un accès antérieur avec sternotomie oblique jusqu'au troisième espace intercostal est réalisé (les vaisseaux sont soigneusement déplacés vers le bas). Si la tumeur affecte les corps de 3 à 5 vertèbres thoraciques, un accès antérolatéral avec résection de la troisième côte est réalisé. L'omoplate est reculée sans sectionner la musculature. Si l'ostéoblastoclastome est localisé dans la région thoracolombaire entre Th11 et L2, l'intervention de choix est une thoracofrénolombotomie droite. L'accès à la partie antérieure des 3 vertèbres supérieures du sacrum est plus difficile. Un accès rétropéritonéal antérolatéral droit avec drainage soigneux des troncs vasculaires et de l'uretère est recommandé.
- Si les corps vertébraux sont gravement détruits ou si la pathologie s'est propagée à la région de la voûte plantaire de la colonne thoracique et lombo-sacrée, dans ce cas, une fixation transpédiculaire-translaminaire de la colonne vertébrale est réalisée, après quoi les vertèbres détruites sont retirées avec une autoplastie supplémentaire.
- Si une forme bénigne d'ostéoblastoclastome est détectée au niveau de l'arcade sourcilière et du sciatique, le segment pathologiquement altéré est retiré dans les tissus sains, sans greffe osseuse. Si le plancher et le toit de l'acétabulum sont touchés, une résection est réalisée avec greffe osseuse complémentaire pour remplacer le défaut, avec fixation par des attaches spongieuses.
- Si l'os iliaque, le sein ou le sciatique est touché, une alloplastie avec une allogreffe structurelle, une ostéosynthèse de transplantation, une insertion plastique à base de ciment et un repositionnement de la tête prothétique dans une cavité artificielle sont réalisés.
- Si le sacrum et L2 sont touchés, une intervention en deux étapes est réalisée, comprenant la résection d'accès postérieur du fragment sacré inférieur pathologiquement altéré (jusqu'à S2 ), la fixation transpédiculaire et l'ablation du néoplasme du côté antérieur par méthode rétropéritonéale avec greffe osseuse.
Dans chaque situation spécifique, le médecin détermine la méthode d’intervention chirurgicale la plus appropriée, en considérant notamment la possibilité d’appliquer les dernières technologies pour améliorer les résultats du traitement et assurer la qualité de vie normale du patient.
La prévention
Il n'existe pas de prévention spécifique de l'ostéoblastoclastome. Cela est dû principalement au manque d'études sur les causes du développement de ces tumeurs. De nombreux experts soulignent la prévention des traumatismes osseux parmi les principaux points de prévention. Cependant, rien n'indique l'influence directe des traumatismes sur la formation de néoplasies osseuses. Dans de nombreux cas, les traumatismes ne font qu'attirer l'attention sur le processus tumoral existant et n'ont pas d'influence évidente sur l'origine du foyer pathologique, mais peuvent contribuer à sa croissance.
Il ne faut pas oublier que l'ostéoblastoclastome se forme souvent dans des os ayant déjà été exposés à des rayonnements ionisants, par exemple pour traiter d'autres tumeurs. Les néoplasmes radio-induits apparaissent généralement au plus tôt trois ans après l'exposition aux rayonnements.
Les mesures préventives non spécifiques comprennent:
- Élimination des mauvaises habitudes;
- Mener un mode de vie sain;
- Nutrition de qualité et durable;
- Activité physique régulière modérée;
- Prévention des blessures, traitement rapide de tout processus pathologique dans le corps, stabilisation de l'immunité.
Prévoir
Les fractures pathologiques surviennent souvent dans la zone osseuse affectée. Dans ce cas, les néoplasies bénignes, sous réserve d'un traitement radical, ont un pronostic favorable, même si la possibilité de récidives et d'une malignité du foyer pathologique n'est pas exclue. Une évolution défavorable de l'ostéoblastoclastome bénin n'est pas exclue si le foyer est caractérisé par une croissance active et une destruction osseuse prononcée. Une telle tumeur peut rapidement détruire un segment osseux entier, entraînant le développement d'une fracture pathologique et une altération significative de la fonction osseuse. Ces patients présentent souvent des difficultés lors du remplacement chirurgical du défaut osseux, et des complications liées à la non-cicatrisation de la fracture apparaissent.
Le taux de survie moyen à cinq ans pour toutes les variantes d'ostéoblastoclastomes malins, tant chez l'enfant que chez l'adulte, est de 70 %, ce qui peut être considéré comme un bon résultat. On peut donc conclure que, dans de nombreux cas, ces néoplasmes sont guéris avec succès. Bien entendu, des facteurs tels que le type de processus tumoral, son stade, le degré de lésion et la malignité sont également importants.
De toute évidence, c'est l'ostéoblastoclastome malin qui représente la plus grande menace. Dans ce cas, on ne peut parler d'un pronostic favorable qu'en cas de détection précoce, de localisation chirurgicale accessible et de sensibilité du foyer aux agents chimiopréventifs et à la radiothérapie.