Expert médical de l'article

Nouvelles publications
Prions - agents responsables des maladies à prions
Dernière revue: 06.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Les infections virales lentes sont caractérisées par des critères particuliers:
- une période d’incubation inhabituellement longue (mois, années);
- une lésion spécifique des organes et des tissus, principalement du système nerveux central;
- progression lente et régulière de la maladie;
- issue fatale inévitable.

Certains agents pathogènes responsables d'infections virales aiguës peuvent également provoquer des infections virales lentes. Par exemple, le virus de la rougeole provoque parfois une PESS, et le virus de la rubéole provoque une rubéole congénitale progressive et une panencéphalite rubéoleuse.
Une infection virale lente typique des animaux est causée par le virus Visna/Madi, un rétrovirus. Il est à l'origine d'une infection virale lente et d'une pneumonie progressive chez les moutons. La substance blanche du cerveau est détruite, une paralysie se développe (visna – dépérissement); une inflammation chronique des poumons et de la rate apparaît.
Les maladies similaires aux infections virales lentes sont causées par les prions, agents responsables des infections à prions. Les maladies à prions regroupent des troubles progressifs du système nerveux central chez l'homme et l'animal. Chez l'homme, le fonctionnement du système nerveux central est altéré, des changements de personnalité et des troubles moteurs apparaissent. Les symptômes de la maladie durent généralement de quelques mois à plusieurs années et se terminent par le décès. Auparavant, les infections à prions étaient considérées comme des agents responsables des infections virales lentes.
Certains agents responsables des maladies à prions s'accumulent d'abord dans les tissus lymphoïdes. Une fois dans le cerveau, les prions s'accumulent en grande quantité, provoquant une amylose (dysprotéinose extracellulaire, caractérisée par le dépôt d'amyloïde avec développement d'une atrophie et d'une sclérose des tissus) et une astrocytose (prolifération de la névroglie astrocytaire, hyperproduction de fibres gliales). Des fibrilles, des agrégats de protéines ou d'amyloïdes et des modifications spongiformes cérébrales (encéphalopathies spongiformes transmissibles) se forment. Il en résulte des modifications du comportement, une altération de la coordination des mouvements et un épuisement potentiellement mortel. L'immunité est absente. Les maladies à prions sont des maladies conformationnelles qui se développent suite à un repliement incorrect (violation de la conformation correcte) des protéines cellulaires nécessaires au fonctionnement normal de l'organisme. Les voies de transmission des prions sont variées:
- voie alimentaire - produits infectés d'origine animale, additifs alimentaires issus d'organes bovins crus, etc.:
- transmission par transfusion sanguine, administration de médicaments d’origine animale, transplantation d’organes et de tissus, utilisation d’instruments chirurgicaux et dentaires infectés;
- transmission par des préparations immunobiologiques (on connaît une infection de 1500 moutons par la PrP''' par le vaccin au formol cérébral provenant de moutons malades).
Les prions pathologiques, une fois entrés dans l'intestin, sont transportés dans le sang et la lymphe. Après réplication périphérique dans la rate, l'appendice, les amygdales et d'autres tissus lymphoïdes, ils sont transférés au cerveau par les nerfs périphériques (neuroinvasion). Une pénétration directe des prions dans le cerveau à travers la barrière hémato-encéphalique est possible. On pensait auparavant que le système nerveux central était le seul tissu où les prions pathologiques s'accumulaient, mais des études ont infirmé cette hypothèse. Il s'est avéré que l'accumulation de prions dans la rate est associée à la croissance et au fonctionnement des cellules dendritiques folliculaires.
 [
[ Propriétés des prions
L'isoforme cellulaire normale de la protéine prion, d'un poids moléculaire de 33 à 35 kDa, est déterminée par le gène de la protéine prion (le gène du prion – PrNP – est situé sur le 20e chromosome humain). Ce gène normal apparaît à la surface cellulaire (ancré dans la membrane par la glycoprotéine de la molécule), et est sensible à la protéase. Il régule la transmission de l'influx nerveux, les cycles quotidiens et les processus d'oxydation, participe au métabolisme du cuivre dans le système nerveux central et à la régulation de la division des cellules souches de la moelle osseuse. De plus, le gène du prion est présent dans la rate, les ganglions lymphatiques, la peau, le tractus gastro-intestinal et les cellules dendritiques folliculaires.
Prolifération de prions pathologiques
La transformation des prions en formes altérées se produit lorsque l'équilibre cinétique entre eux est perturbé. Ce processus est favorisé par une augmentation de la quantité de prion pathologique (PrP) ou exogène. La PrP est une protéine normale ancrée dans la membrane cellulaire. La PrP' est une protéine hydrophobe globulaire qui forme des agrégats avec elle-même et la PrP'' à la surface cellulaire: la PrP' se transforme alors en PrP'', et le cycle se poursuit. La forme pathologique de la PrP''' s'accumule dans les neurones, donnant à la cellule un aspect spongieux.
Kuru
La maladie à prions (ou maladie à prions), autrefois courante chez les Papous (terme qui signifie « tremblements » ou « tremblements ») de l'est de l'île de Nouvelle-Guinée, a été démontrée par K. Gajdusek. L'agent pathogène se transmet par voie alimentaire lors d'un cannibalisme rituel: la consommation du cerveau insuffisamment cuit et infecté par le prion de proches décédés. Suite à des lésions du système nerveux central, les mouvements et la démarche sont perturbés, des frissons et une euphorie (« mort rieuse ») apparaissent. La période d'incubation dure de 5 à 30 ans. Le patient décède au bout d'un an.
Maladie de Creutzfeldt-Jakob
Maladie à prions, qui se manifeste par une démence, des troubles visuels et cérébelleux et des troubles du mouvement, avec une issue fatale après 4 à 5 mois de maladie dans la variante classique de la maladie de Creutzfeldt-Jakob et après 3 à 14 mois dans la nouvelle variante de la maladie. La période d'incubation peut atteindre 20 ans. Différentes voies d'infection et causes de la maladie sont possibles:
- lors de la consommation de produits animaux insuffisamment traités thermiquement, tels que la viande et la cervelle de vaches atteintes d’encéphalopathie spongiforme bovine;
- lors de transplantations de tissus, telles que la transplantation de cornée, la transfusion sanguine, l'utilisation d'hormones et d'autres substances biologiquement actives d'origine animale, l'utilisation de catgut, d'instruments chirurgicaux contaminés ou insuffisamment stérilisés, les manipulations prosectorielles;
- en cas d'hyperproduction de PrR et d'autres conditions qui stimulent le processus de conversion de PrR' en PrR".
La maladie peut également se développer suite à une mutation ou à une insertion dans la région du gène prion. Le caractère familial de la maladie est fréquent en raison d'une prédisposition génétique à la maladie de Creutzfeldt-Jakob. Dans la nouvelle variante de la maladie de Creutzfeldt-Jakob, les troubles se développent à un âge plus jeune (âge moyen 28 ans), contrairement à la variante classique (âge moyen 65 ans). Dans cette nouvelle variante, une protéine prion anormale s'accumule non seulement dans le système nerveux central, mais aussi dans les tissus lymphoréticulaires, notamment les amygdales.
Syndrome de Gerstmann-Sträussler-Scheinker
Maladie à prions héréditaire, accompagnée de démence, d'hypotonie, de troubles de la déglutition (dysphagie) et de dysarthrie. Elle est souvent de nature familiale. La période d'incubation est de 5 à 30 ans. La maladie survient entre 50 et 60 ans et dure de 5 à 13 ans.
Insomnie mortelle héréditaire
Maladie auto-immune caractérisée par une insomnie progressive, une hyperréactivité sympathique (hypertension, hyperthermie, hyperhidrose, tachycardie), des tremblements, une ataxie, une multiclonie et des hallucinations. Le sommeil est gravement perturbé. La mort survient avec la progression de l'insuffisance cardiovasculaire.
Gratter
La tremblante (de l'anglais scrape - gratter) est une maladie à prions des moutons et des chèvres (gale), qui se manifeste par des lésions du système nerveux central, des troubles progressifs du mouvement, de fortes démangeaisons cutanées (gale) et se termine par la mort de l'animal.
Encéphalopathie spongiforme bovine
Maladie du bétail caractérisée par des lésions du système nerveux central, une altération de la coordination des mouvements et la mort inévitable de l'animal. L'épidémie s'est d'abord déclarée en Grande-Bretagne. Elle était associée à l'alimentation des animaux avec des farines de viande et d'os contenant des prions pathogènes. La période d'incubation varie de 1,5 à 15 ans. Le cerveau, la moelle épinière et les globes oculaires sont les plus infectés.
Diagnostic en laboratoire des maladies à prions
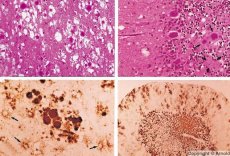
Lors du diagnostic, des modifications spongiformes cérébrales, une astrocytose (gliose) et l'absence d'infiltrats inflammatoires sont observées. Le cerveau est coloré à la recherche d'amyloïde. Des marqueurs protéiques des maladies cérébrales à prions sont détectés dans le liquide céphalorachidien (par ELISA). Une analyse génétique du gène du prion (PCR) est réalisée.
Prévention des maladies à prions
Pour la décontamination des instruments et des objets environnementaux, il est recommandé d'autoclaver (à 134 °C pendant 18 min; à 121 °C pendant 1 h), d'incinérer, puis de traiter les instruments et les objets environnementaux avec de l'eau de Javel et une solution de NaCl à un pH pendant 1 h. Pour la prophylaxie non spécifique, des restrictions ont été introduites concernant l'utilisation de médicaments d'origine animale et la production d'hormones hypophysaires d'origine animale est interdite. La transplantation de dure-mère est interdite. Le port de gants en caoutchouc est obligatoire pour manipuler les liquides physiologiques des patients.