Expert médical de l'article

Nouvelles publications
Mucoviscidose
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
La fibrose kystique est une maladie monogénique génétique autosomique récessive caractérisée par un trouble de la sécrétion des glandes exocrines des organes vitaux avec atteinte principalement des systèmes respiratoire et digestif, évolution sévère et pronostic défavorable.
 [ 1 ]
[ 1 ]
Épidémiologie
L'incidence de la mucoviscidose varie entre 1/2 500 et 1/4 600 nouveau-nés. Chaque année, environ 45 000 personnes atteintes de mucoviscidose naissent dans le monde. L'incidence des porteurs du gène de la mucoviscidose est de 3 à 4 %, avec environ 275 millions de personnes dans le monde porteuses de ce gène, dont environ 5 millions vivent en Russie et environ 12,5 millions dans les pays de la CEI.
Causes mucoviscidose
La mucoviscidose se transmet selon un mode autosomique récessif. Le gène responsable de la mucoviscidose est situé dans l'autosome 7, contient 27 exons et est constitué de 250 000 paires de nucléotides.
Un même gène peut présenter de nombreuses mutations, chacune spécifique à une population ou une région géographique particulière. Plus de 520 mutations ont été décrites, la plus fréquente étant la mutation delta-P-508, c'est-à-dire une substitution de l'acide aminé phénylalanine en position 508.
Pathogénèse
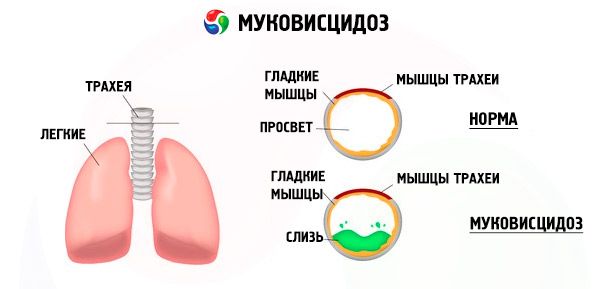
Des mutations du gène de la mucoviscidose perturbent la structure et la fonction d'une protéine appelée CFTR (cystic fibrosis transmembrane regulator). Cette protéine agit comme un canal chlorure impliqué dans les échanges eau-électrolytes des cellules épithéliales du système bronchopulmonaire, du tube digestif, du pancréas, du foie et de l'appareil reproducteur. Suite à cette perturbation, les ions chlorure Cl- s'accumulent à l'intérieur de la cellule. Ceci entraîne une modification du potentiel électrique dans la lumière des canaux excréteurs, ce qui facilite l'écoulement de grandes quantités d'ions sodium (Na + ) de la lumière du canal vers la cellule et favorise l'absorption d'eau depuis l'espace péricellulaire.
À la suite de ces changements, la sécrétion de la plupart des glandes exocrines s'épaissit, son évacuation est perturbée, ce qui entraîne des troubles secondaires prononcés dans les organes et les systèmes, plus prononcés dans les systèmes bronchopulmonaire et digestif.
Un processus inflammatoire chronique d'intensité variable se développe dans les bronches, la fonction de l'épithélium cilié est fortement perturbée, les expectorations deviennent très visqueuses, épaisses et difficiles à évacuer, et une stagnation est observée. Des bronchiolo- et des bronchectasies se forment, qui deviennent plus fréquentes avec le temps. Ces changements entraînent une augmentation de l'hypoxie et la formation d'une maladie pulmonaire chronique.
Les patients atteints de mucoviscidose sont extrêmement prédisposés au développement d'une inflammation chronique du système bronchopulmonaire. Ceci est dû à des troubles prononcés du système de défense bronchopulmonaire local (diminution des taux d'IgA, d'interféron, de la fonction phagocytaire des macrophages alvéolaires et des leucocytes).
Les macrophages alvéolaires jouent un rôle majeur dans le développement de l'inflammation chronique du système bronchopulmonaire. Ils produisent de grandes quantités d'IL-8, ce qui augmente considérablement la chimiotaxie des neutrophiles dans l'arbre bronchique. Les neutrophiles s'accumulent en grande quantité dans les bronches et, avec les cellules épithéliales, sécrètent de nombreuses cytokines pro-inflammatoires, notamment l'IL-1, l'IL-8 et l'IL-6, le facteur de nécrose tumorale et les leucotriènes.
L'activité élevée de l'enzyme élastase joue également un rôle important dans la pathogenèse des lésions du système bronchopulmonaire. On distingue l'élastase exogène de l'élastase endogène. La première est produite par la flore bactérienne (notamment Pseudomonas aeruginosa), la seconde par les leucocytes neutrophiles. L'élastase détruit l'épithélium et d'autres éléments structurels des bronches, ce qui contribue à perturber davantage le transport mucociliaire et à la formation rapide de bronchectasies.
Les leucocytes neutrophiles sécrètent également d'autres enzymes protéolytiques. L'alpha-1-antipyrsine et l'inhibiteur sécrétoire des leucoprotéases neutralisent l'influence des enzymes protéolytiques et protègent ainsi le système bronchopulmonaire de leurs effets néfastes. Malheureusement, chez les patients atteints de mucoviscidose, ces facteurs protecteurs sont supprimés par une quantité importante de protéase neutrophile.
Toutes ces circonstances contribuent à l'introduction de l'infection dans le système bronchopulmonaire et au développement d'une bronchite chronique purulente. De plus, il convient de prendre en compte que la protéine défectueuse codée par le gène de la mucoviscidose modifie l'état fonctionnel de l'épithélium bronchique, favorisant ainsi l'adhésion de bactéries, principalement Pseudomonas aeruginosa.
Outre la pathologie du système bronchopulmonaire, la fibrose kystique provoque également de graves lésions du pancréas, de l’estomac, du gros et du petit intestin et du foie.
Symptômes mucoviscidose
La mucoviscidose se manifeste par divers symptômes cliniques. Chez le nouveau-né, la maladie peut se manifester par un iléus méconial. En raison d'un déficit, voire d'une absence totale, de trypsine, le méconium devient très dense et visqueux et s'accumule dans la région iléo-cæcale. Une occlusion intestinale se développe ensuite, se manifestant par des vomissements intenses avec imprégnation biliaire, une distension abdominale, une absence d'excrétion méconiale, l'apparition de symptômes de péritonite et une aggravation rapide des manifestations cliniques du syndrome d'intoxication sévère. L'enfant peut décéder dès les premiers jours de vie en l'absence d'intervention chirurgicale urgente.
Dans les cas moins graves, un signe caractéristique de la mucoviscidose est une selle abondante, fréquente, onctueuse, riche en graisses et d'odeur très désagréable. Un prolapsus rectal est observé chez un tiers des patients.
Par la suite, les patients continuent de souffrir de dysfonctionnement intestinal, de syndrome de malabsorption, de troubles graves du développement physique et d’hypovitaminose sévère.
Au cours de la première ou de la deuxième année de vie, des symptômes d'atteinte du système bronchopulmonaire (forme bénigne de la maladie) apparaissent, se manifestant par une toux parfois très prononcée et ressemblant à une toux coquelucheuse. Cette toux s'accompagne d'une cyanose, d'un essoufflement et de la production d'expectorations épaisses, d'abord muqueuses, puis purulentes. Progressivement, un tableau clinique de bronchite chronique obstructive et de bronchectasie, d'emphysème pulmonaire et d'insuffisance respiratoire se développe. Les enfants sont extrêmement sensibles aux infections respiratoires aiguës virales et bactériennes, ce qui contribue à l'exacerbation et à la progression de la pathologie bronchopulmonaire. Le développement d'un asthme bronchique infectieux est possible.
Chez les enfants d'âge scolaire, la mucoviscidose peut se manifester par des coliques intestinales. Les patients se plaignent de douleurs abdominales paroxystiques intenses, de ballonnements et de vomissements répétés. La palpation abdominale révèle des formations denses situées dans la projection du gros intestin: des masses fécales mêlées à un mucus épais et dense. Les enfants sont très sujets au développement d'une alcalose hypochlorémique due à l'excrétion excessive de sel par la transpiration par temps chaud, tandis que des « gelées de sel » apparaissent sur la peau.
Affections du système bronchopulmonaire chez l'adulte
Les lésions du système bronchopulmonaire chez les patients atteints de mucoviscidose (forme pulmonaire de la maladie) se caractérisent par l'apparition d'une bronchite chronique purulente obstructive, de bronchectasies, d'une pneumonie chronique, d'un emphysème pulmonaire, d'une insuffisance respiratoire et d'une cardiopathie pulmonaire. Certains patients développent un pneumothorax et d'autres complications de la mucoviscidose: atélectasie, abcès pulmonaires, hémoptysie, hémorragie pulmonaire et asthme bronchique infectieux.
Les patients se plaignent d'une toux paroxystique douloureuse accompagnée d'expectorations mucopurulentes très visqueuses et difficiles à séparer, parfois mêlées de sang. De plus, un essoufflement est très caractéristique, d'abord à l'effort physique, puis au repos. Cet essoufflement est dû à une obstruction bronchique. De nombreux patients se plaignent de rhinite chronique due à une polypose et une sinusite. Une faiblesse importante, une baisse progressive des performances et des infections virales respiratoires aiguës fréquentes sont également caractéristiques. À l'examen, on remarque une pâleur de la peau, un gonflement du visage, une cyanose des muqueuses visibles et un essoufflement sévère. Avec le développement d'une cardiopathie pulmonaire décompensée, un œdème apparaît au niveau des jambes. On peut observer un épaississement des phalanges terminales des doigts en forme de pilons et des ongles en forme de verre de montre. Le thorax prend une forme de tonneau (due au développement d'un emphysème pulmonaire).
La percussion pulmonaire révèle des signes d'emphysème: un bruit de caisse, une limitation marquée de la mobilité du bord pulmonaire et un abaissement du bord inférieur des poumons. L'auscultation pulmonaire révèle une respiration difficile avec une expiration prolongée, des sifflements secs dispersés et des sifflements humides, moyens et fins, avec des bulles. En cas d'emphysème pulmonaire sévère, la respiration est fortement affaiblie.
Manifestations extrapulmonaires de la mucoviscidose
Les manifestations extrapulmonaires de la fibrose kystique peuvent être assez prononcées et survenir fréquemment.
Lésions pancréatiques
Une insuffisance de la fonction exocrine du pancréas, plus ou moins grave, est observée chez 85 % des patients atteints de mucoviscidose. En cas de lésions pancréatiques mineures, les syndromes de maldigestion et de malabsorption sont absents; seuls des signes biologiques d'insuffisance exocrine sont observés (faibles taux de trypsine et de lipase dans le sang et le contenu duodénal; stéatorrhée souvent sévère). Il est connu que pour prévenir le syndrome de maldigestion, la sécrétion de seulement 1 à 2 % de la lipase totale suffit. Seules les perturbations significatives de la fonction exocrine se manifestent cliniquement.
En conditions normales, les acini du pancréas produisent une sécrétion liquide riche en enzymes. En parcourant le canal excréteur, la sécrétion s'enrichit en eau et en anions, devenant encore plus liquide. Dans la mucoviscidose, en raison d'un trouble de la structure et de la fonction du régulateur transmembranaire (canal chlorure), la sécrétion pancréatique ne reçoit pas suffisamment de liquide, devient visqueuse et sa vitesse de déplacement le long du canal excréteur ralentit fortement. Les protéines de la sécrétion se déposent sur les parois des petits canaux excréteurs, entraînant leur obstruction. À mesure que la maladie progresse, la destruction et l'atrophie des acini finissent par se développer: une pancréatite chronique avec insuffisance pancréatique exocrine se développe. Ceci se traduit cliniquement par le développement de syndromes de maldigestion et de malabsorption. L'insuffisance pancréatique est la principale cause de malabsorption des graisses dans la mucoviscidose, mais elle est généralement observée en cas de déficit important en lipase. Forsher et Durie (1991) indiquent qu'en l'absence totale de lipase pancréatique, les graisses sont dégradées et absorbées à 50-60 %, ce qui est dû à la présence de lipases gastriques et salivaires (sublinguales), dont l'activité est proche de la limite inférieure de la norme. Parallèlement à la perturbation de la dégradation et de l'absorption des graisses, on observe une perturbation de la dégradation et de la réabsorption des protéines. Environ 50 % des protéines absorbées par l'alimentation sont éliminées dans les selles. L'absorption des glucides est moins affectée malgré le déficit en α-amylase, mais le métabolisme des glucides peut être significativement perturbé.
Les lésions du pancréas se traduisent par le développement d'un syndrome de maldigestion et de malabsorption avec perte de poids importante et selles grasses abondantes.
Le développement de syndromes de maldigestion et de malabsorption est également facilité par un dysfonctionnement grave des glandes intestinales, une altération de la sécrétion du suc intestinal et une diminution de la teneur en enzymes intestinales.
Les syndromes de maldigestion et de malabsorption sont également appelés forme intestinale de la fibrose kystique.
Une altération de la fonction endocrine du pancréas (diabète sucré) est observée chez les patients atteints de mucoviscidose aux stades avancés de la maladie (chez 2 % des enfants et 15 % des adultes).
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Lésions du foie et des voies biliaires
Chez 13 % des patients atteints de mucoviscidose mixte et intestinale, une cirrhose hépatique se développe. Elle est plus fréquente avec les mutations W128X, delta-P508 et X1303K. Une cirrhose biliaire avec hypertension portale est détectée chez 5 à 10 % des patients. Selon Welch et Smith (1995), des signes cliniques, morphologiques, biologiques et instrumentaux d'atteinte hépatique sont détectés chez 86 % des patients atteints de mucoviscidose.
De nombreux patients atteints de mucoviscidose développent également une cholécystite chronique, souvent calculeuse.
Dysfonctionnement des glandes sexuelles
Les patients atteints de mucoviscidose peuvent souffrir d'azoospermie, cause d'infertilité. Une baisse de fertilité est également fréquente chez les femmes.
Étapes
Il existe trois degrés de gravité de la mucoviscidose pulmonaire.
La forme légère de la fibrose kystique se caractérise par de rares exacerbations (pas plus d'une fois par an); pendant les périodes de rémission, les manifestations cliniques sont pratiquement absentes et les patients sont capables de travailler.
Gravité modérée: les exacerbations sont observées 2 à 3 fois par an et durent environ 2 mois, voire plus. En phase d'exacerbation, on observe une toux intense avec expectorations difficiles à séparer, un essoufflement même après un effort physique léger, une fièvre légère, une faiblesse générale et des sueurs. Parallèlement, on observe une altération de la fonction exocrine du pancréas. En phase de rémission, la capacité de travail n'est pas totalement rétablie et l'essoufflement persiste à l'effort physique.
L'évolution sévère se caractérise par des exacerbations très fréquentes de la maladie. Les rémissions sont pratiquement absentes. Le tableau clinique met en évidence une insuffisance respiratoire sévère, des symptômes de cardiopathie pulmonaire chronique, souvent décompensés, et une hémoptysie typique. Une perte de poids importante est observée, et les patients sont totalement invalides. En règle générale, une pathologie bronchopulmonaire sévère s'accompagne d'un dysfonctionnement pancréatique prononcé.
Formes
- Lésions bronchopulmonaires
- Pneumonie répétée et récurrente avec une évolution prolongée.
- Pneumonie abcédée, en particulier chez les nourrissons.
- Pneumonie chronique, notamment bilatérale.
- Asthme bronchique réfractaire au traitement traditionnel.
- Bronchite récidivante, bronchiolite, notamment avec culture de Pseudomonas aeruginosa.
- Modifications du tractus gastro-intestinal
- Iléus méconial et ses équivalents.
- Syndrome d'altération de l'absorption intestinale d'origine inconnue.
- Ictère obstructif du nouveau-né avec une évolution prolongée.
- Cirrhose du foie.
- Diabète sucré.
- Reflux gastro-oesophagien.
- Lithiase biliaire.
- Prolapsus rectal.
- Modifications dans d'autres organes et systèmes
- Troubles de la croissance et du développement.
- Retard du développement sexuel.
- Infertilité masculine.
- Polypes nasaux.
- Frères et sœurs issus de familles atteintes de fibrose kystique.
[ 24 ]
Complications et conséquences
Les complications du tractus gastro-intestinal comprennent:
- Le diabète sucré se développe chez 8 à 12 % des patients de plus de 25 ans.
- Colonopathie fibrosante.
- Iléus méconial en période néonatale (chez 12 % des nouveau-nés atteints de mucoviscidose), syndrome d'obstruction intestinale distale, prolapsus rectal, ulcère gastroduodénal et reflux gastro-œsophagien.
Complications hépatiques:
- Stéatose hépatique (chez 30 à 60 % des patients),
- Cirrhose biliaire focale, cirrhose biliaire multinodulaire et hypertension portale associée.
L'hypertension portale conduit parfois à la mort en raison de varices œsophagiennes.
La prévalence de la cholécystite et des calculs biliaires est plus élevée chez les patients atteints de fibrose kystique que chez les autres individus.
Retard de puberté, baisse de fertilité et autres complications. La plupart des hommes souffrent d'azoospermie et d'un sous-développement des canaux déférents.
Diagnostics mucoviscidose
Analyse sanguine générale: une anémie de gravité variable est typique, généralement normochrome ou hypochrome. L'anémie est d'origine polyfactorielle (diminution de l'absorption du fer et de la vitamine B12 dans l'intestin due au développement d'un syndrome de malabsorption). Une leucopénie est possible, avec développement d'une bronchite purulente et d'une pneumonie (leucocytose, augmentation de la VS).
Analyse d'urine générale - aucun changement significatif, dans de rares cas une légère protéinurie est observée.
Examen coprologique: stéatorrhée et créatorrhée sont observées. Becker (1987) recommande de doser la chymotrypsine et les acides gras dans les selles. Avant de doser la chymotrypsine dans les selles, il est nécessaire d’arrêter la prise d’enzymes digestives au moins 3 jours avant l’examen. Dans la mucoviscidose, la quantité de chymotrypsine dans les selles est réduite et celle d’acides gras est augmentée (l’excrétion normale d’acides gras est inférieure à 20 mmol/jour). Il est important de noter qu’une excrétion accrue d’acides gras dans les selles est également observée chez:
- déficit en acides gras conjugués dans l'intestin grêle en raison d'une insuffisance hépatique, d'une obstruction des voies biliaires, d'une colonisation bactérienne importante de l'intestin grêle (dans ce cas, une hydrolyse intensive des acides biliaires se produit);
- iléite;
- maladie cœliaque (avec développement d’un syndrome de malabsorption);
- entérite;
- lymphomes intestinaux;
- maladie de Whipple;
- allergies alimentaires;
- transit accéléré des masses alimentaires dans les diarrhées d'origines diverses, syndrome carcinoïde, thyrotoxicose.
Analyse biochimique du sang - diminution des taux de protéines totales et d'albumine, augmentation des taux d'alpha2 et de gamma globulines, de bilirubine et d'aminotransférases (en cas de lésions hépatiques), diminution de l'activité de l'amylase, de la lipase, de la trypsine et des taux de fer et de calcium (en cas de développement d'un syndrome de maldigestion, de malabsorption).
Analyse des expectorations - présence d'un grand nombre de leucocytes neutrophiles et de micro-organismes (lors de la bactérioscopie des expectorations).
L'étude de la fonction d'absorption de l'intestin grêle et de la fonction exocrine du pancréas révèle des perturbations importantes.
L'examen radiographique des poumons révèle des modifications dont la gravité dépend de la gravité et du stade de la maladie. Les modifications les plus caractéristiques sont:
- augmentation du schéma pulmonaire en raison de modifications interstitielles péribronchiques;
- expansion des racines des poumons;
- image d'atélectasie lobulaire, sous-segmentaire ou même segmentaire des poumons;
- transparence accrue des champs pulmonaires, principalement dans les sections supérieures, position basse et mobilité insuffisante du diaphragme, expansion de l'espace rétrosternal (manifestation d'un emphysème pulmonaire);
- infiltration segmentaire ou polysegmentaire du tissu pulmonaire (dans le développement d'une pneumonie).
La bronchographie révèle des modifications causées par l'obstruction des bronches par des expectorations visqueuses (fragmentation du remplissage des bronches avec contraste, contours irréguliers, phénomène de rupture bronchique, diminution significative du nombre de branches latérales), ainsi que des bronchoecgases (cylindriques, mixtes), localisées principalement dans les parties inférieures des poumons.
La bronchoscopie révèle une bronchite purulente diffuse avec des expectorations abondantes, épaisses et visqueuses et des films fibrineux.
Spirométrie - déjà aux premiers stades de la maladie révèle une insuffisance respiratoire de type obstructive (diminution de la CVF, du VEMS, de l'indice de Tiffno), restrictive (diminution de la CVF) ou, le plus souvent, obstructive-restrictive (diminution de la CVF, du VEMS, de l'indice de Tiffno).
Le test de la sueur de Gibson et Cook (test électrolytique de la sueur) consiste à stimuler la sudation par électrophorèse à la pilocarpine, puis à doser les chlorures dans la sueur. Doerehuk (1987) décrit ce test comme suit: l’électrophorèse à la pilocarpine est réalisée sur l’avant-bras, le courant électrique étant de 3 mA. Après avoir nettoyé la peau à l’eau distillée, la sueur est recueillie à l’aide d’un papier filtre placé sur la zone stimulée, recouvert d’une gaze pour éviter son évaporation. Après 30 à 60 minutes, le papier filtre est retiré et élué dans de l’eau distillée. La quantité de sueur recueillie est mesurée. Pour obtenir des résultats fiables, il est nécessaire de recueillir au moins 50 mg (de préférence 100 mg) de sueur.
Si la concentration en chlorure est supérieure à 60 mmol/l, le diagnostic de mucoviscidose est considéré comme probable; si la concentration en chlorure est supérieure à 100 mmol/l, il est fiable; dans ce cas, la différence entre les concentrations de chlore et de sodium ne doit pas dépasser 8 à 10 mmol/l. Hadson (1983) recommande, si la teneur en sodium et en chlorure dans la sueur est limite, de réaliser un test à la prednisolone (5 mg par voie orale pendant 2 jours, suivi d'un dosage des électrolytes dans la sueur). Chez les personnes non atteintes de mucoviscidose, le taux de sodium dans la sueur diminue jusqu'à la limite inférieure de la normale; en cas de mucoviscidose, il ne change pas. Un test de la sueur est recommandé pour tout enfant souffrant de toux chronique.
L'analyse de taches de sang ou d'échantillons d'ADN pour détecter les mutations majeures du gène de la mucoviscidose est le test diagnostique le plus sensible et le plus spécifique. Cependant, cette méthode ne convient qu'aux pays où le taux de mutation delta-P508 est supérieur à 80 %. De plus, cette technique est très coûteuse et techniquement complexe.
Le diagnostic prénatal de la mucoviscidose est réalisé par dosage des isoenzymes de la phosphatase alcaline dans le liquide amniotique. Cette méthode est possible entre 18 et 20 semaines de grossesse.
Les principaux critères de diagnostic de la mucoviscidose sont les suivants:
- indications dans l'anamnèse d'un retard de développement physique dans l'enfance, de maladies respiratoires chroniques récurrentes, de troubles dyspeptiques et de diarrhée, de présence de mucoviscidose chez des parents proches;
- bronchite chronique obstructive, souvent récidivante, avec développement de bronchectasies et d'emphysème pulmonaire, pneumonie souvent récidivante;
- pancréatite chronique récurrente avec diminution marquée de la fonction exocrine, syndrome de malabsorption;
- augmentation de la teneur en chlore dans la sueur du patient;
- infertilité avec fonction sexuelle préservée.
Le diagnostic réussi et le diagnostic différentiel de la fibrose kystique sont facilités par l’identification des groupes à risque.
Programme de dépistage de la fibrose kystique
- Analyse générale de sang, d'urine, d'expectorations.
- Analyse bactériologique des expectorations.
- Analyse coprologique.
- Analyse biochimique du sang: détermination des protéines totales et des fractions protéiques, du glucose, de la bilirubine, des aminotransférases, de la phosphatase alcaline, des gamma-glutamyl transpeptidases, du potassium, du calcium, du fer, de la lipase, de l'amylase, de la trypsine.
- Étude de la fonction exocrine du pancréas et de la fonction absorbante de l'intestin.
- Fluoroscopie et radiographie des poumons, scanner des poumons.
- ECG.
- Échocardiographie.
- Bronchoscopie et bronchographie.
- Spirométrie.
- Test de sueur.
- Consultation avec un généticien.
- Analyse de taches de sang ou d'échantillons d'ADN pour détecter les mutations majeures du gène de la fibrose kystique.
Comment examiner?
Quels tests sont nécessaires?
Qui contacter?
Traitement mucoviscidose
Le type et la gravité des symptômes de la fibrose kystique peuvent varier considérablement, il n’existe donc pas de plan de traitement typique; il est individualisé pour chaque individu.
La thérapie comprend les mesures thérapeutiques suivantes:
- Les exercices de respiration et le drainage postural aident à éliminer le mucus épais qui s'accumule dans les poumons. Certaines techniques de dégagement des voies respiratoires nécessitent l'aide de membres de la famille, d'amis ou d'un pneumologue. De nombreuses personnes utilisent une veste thoracique gonflable qui vibre à haute fréquence.

- Médicaments par inhalation ayant des effets bronchodilatateurs, drainants (mucolytiques) et antibactériens (par exemple, les fluoroquinolones).
- Préparations contenant des enzymes pancréatiques pour améliorer la digestion. Ces préparations sont prises pendant les repas.
- Multivitamines (y compris vitamines liposolubles).
En 2015, la FDA a approuvé un deuxième médicament pour traiter la mucoviscidose, ciblant une protéine défectueuse appelée CFTR. Le premier médicament, un modulateur de CFTR, a été approuvé en 2012. Les modulateurs de CFTR devraient prolonger de plusieurs décennies la vie de certaines personnes atteintes de mucoviscidose.
Une intervention chirurgicale peut être nécessaire pour traiter les complications respiratoires suivantes:
- Pneumothorax, hémoptysie massive récurrente ou persistante, polypes nasaux, sinusite persistante et chronique.
- Iléus méconial, invagination, prolapsus rectal.
La transplantation pulmonaire est réalisée au stade terminal de la maladie.
Prévoir
L'âge moyen de survie des patients atteints de mucoviscidose varie de 35 à 40 ans. Il est plus élevé chez les hommes que chez les femmes.
Grâce aux traitements modernes, 80 % des patients atteignent l'âge adulte. Cependant, la mucoviscidose limite considérablement les capacités fonctionnelles des patients. Il n'existe toujours pas de traitement curatif pour cette maladie.