Expert médical de l'article

Nouvelles publications
Le neuroblastome chez l'enfant: causes, diagnostic, traitement
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
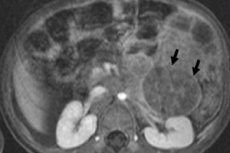
En oncologie pédiatrique, l'un des néoplasmes extracrâniens les plus courants est le neuroblastome chez les enfants, qui est une tumeur maligne embryonnaire des neuroblastes de la crête neurale, c'est-à-dire des cellules nerveuses embryonnaires (immatures) du système nerveux sympathique.
Épidémiologie
Selon les statistiques de l'International Neuroblastoma Risk Group (INRG), le neuroblastome représente environ 8 % de toutes les maladies oncologiques chez les enfants dans le monde et se classe au troisième rang en termes de prévalence, après la leucémie et les tumeurs cérébrales.
Selon d'autres données, le neuroblastome représente environ 28 % de tous les cancers chez les nourrissons. Plus d'un tiers des cas de neuroblastome sont diagnostiqués chez des enfants de moins d'un an; l'âge moyen du diagnostic est de 19 à 22 mois. Plus de 90 % des cas diagnostiqués surviennent chez des enfants âgés de deux à cinq ans (avec une prédominance de garçons); l'incidence maximale est observée entre deux et trois ans, et les cas chez les enfants de plus de cinq ans représentent moins de 10 %.
Causes neuroblastomes
En étudiant les causes du neuroblastome, les chercheurs ont conclu que cette tumeur infantile est due à des mutations génétiques sporadiques survenues au cours de l'embryogenèse ou du développement postnatal précoce. Cependant, les causes de ces modifications génétiques demeurent inconnues, l'influence de facteurs environnementaux tératogènes n'ayant pas été identifiée.
Ces tumeurs peuvent apparaître n’importe où, y compris dans le médiastin, le cou, l’abdomen, les glandes surrénales, les reins, la colonne vertébrale et le bassin.
Dans de rares cas, le neuroblastome du nourrisson peut être associé à une mutation héréditaire. Il s'agit notamment d'une mutation du gène de la protéine membranaire CD246 (chromosome 2) – l'enzyme tyrosine kinase ALK, qui assure la communication intercellulaire et joue un rôle important dans le fonctionnement du système nerveux –; et du gène de la protéine PHOX2B (chromosome 4), impliquée dans la maturation des cellules nerveuses.
Le neuroblastome peut également être associé à la neurofibromatose infantile de type 1,au syndrome de Beckwith-Wiedemann et à l'hypoglycémie hyperinsulinémique (pancréatite à nésidioblastose).
Facteurs de risque
Aujourd'hui, l'hérédité est reconnue comme un facteur de risque de développement du neuroblastome chez l'enfant: la présence de cette tumeur dans les antécédents familiaux, ainsi que les anomalies congénitales associées à des mutations génétiques survenues au cours du développement intra-utérin. Cela est particulièrement vrai en cas de développement de plusieurs néoplasmes dans différents organes.
Aucun des facteurs exogènes augmentant le risque de cette tumeur n’a été identifié par les chercheurs.
Pathogénèse
Le mécanisme de développement des neuroblastomes est dû à des troubles de la différenciation et de la maturation des cellules de la crête neurale – des lignées cellulaires bilatérales qui se forment aux abords du tube neural à partir de la couche embryonnaire ectodermique de l'embryon humain. Ces cellules migrent (se déplacent) et se différencient en de nombreux types cellulaires: neurones sensoriels et autonomes, cellules neuroendocrines et cellules de la médullosurrénale, cellules du cartilage craniofacial et des os, ainsi que cellules pigmentaires.
Dans le neuroblastome, les neuroblastes migrés ne parviennent pas à maturité, mais continuent de croître et de se diviser, formant une tumeur. La pathogenèse de sa formation est associée aux mutations génétiques suivantes:
- avec duplication d'une partie de la séquence chromosomique ou duplication de segments du gène LMO1 sur le chromosome 11, codant pour la protéine RBTN1 dans les cellules de la crête neurale de l'embryon;
- avec une modification du nombre de copies du gène NBPF10 sur le chromosome 1q21.1, codant pour la protéine DUF1220, qui contrôle la prolifération des cellules souches neurales humaines. Ces troubles entraînent soit une duplication de ce chromosome, soit sa délétion (absence d'une partie de l'ADN);
- avec des changements dans le gène suppresseur de tumeur ATRX (sur le chromosome Xq21.1);
- avec la présence de copies supplémentaires (amplification) du gène du facteur de transcription N-Myc sur le chromosome 2, codant pour l'un des facteurs de transcription (protéine de liaison à l'ADN) régulant l'activité d'autres gènes et contrôlant la prolifération des cellules précurseurs lors de la formation des protéines nécessaires à la formation des tissus et organes du fœtus. L'amplification de ce gène le transforme en oncogène, provoquant une perturbation du cycle cellulaire, une prolifération cellulaire accrue et la formation de tumeurs.
Symptômes neuroblastomes
Les premiers signes du neuroblastome ne sont pas spécifiques et peuvent inclure une perte d’appétit (et une perte de poids), de la fatigue lors de l’alimentation, de la fièvre et des douleurs articulaires.
Les symptômes cliniques dépendent de la localisation de la tumeur primaire et de la présence de métastases (qui surviennent dans 60 à 73 % des cas).
Très souvent, le neuroblastome primitif est localisé dans la médullosurrénale, dont l'origine est similaire à celle des cellules nerveuses. Chez les enfants de moins d'un an, le neuroblastome surrénalien est diagnostiqué dans 35 à 40 % des cas. Ses symptômes incluent des douleurs abdominales, de la fièvre, une perte de poids, des douleurs osseuses, une anémie ou un syndrome de Pepper concomitant: atteinte hépatique diffuse avec hépatomégalie sévère et syndrome de détresse respiratoire.
Le neuroblastome rétropéritonéal ou neuroblastome rétropéritonéal chez l'enfant, à mesure qu'il grandit, commence à appuyer sur la vessie ou les intestins, ce qui peut provoquer des problèmes de miction ou de défécation, un gonflement des jambes (chez les garçons, le scrotum gonfle).
Le neuroblastome médiastinal chez l'enfant comprime souvent la veine cave supérieure, ce qui peut provoquer un gonflement du visage, du cou, des bras et de la partie supérieure du thorax (la peau prenant une teinte bleu-rouge, avec des nodules sous-cutanés). Une toux et une respiration sifflante, des difficultés respiratoires (essoufflement) ou des troubles de la déglutition (dysphagie) apparaissent; on observe une hypertrophie des ganglions lymphatiques au niveau du cou, au-dessus de la clavicule et des aisselles.
La propagation des cellules tumorales à la moelle osseuse entraîne une anémie, une thrombocytopénie et une leucopénie avec une tendance au saignement.
En cas de métastases périorbitaires, des cernes ou des ecchymoses apparaissent autour des yeux. Une telle tumeur peut également provoquer des maux de tête et des vertiges, une exophtalmie (gonflement des globes oculaires) et, en raison de la compression des terminaisons nerveuses, un affaissement des paupières (ptosis) et une diminution de la taille des pupilles (myosis).
Le neuroblastome abdominal, ou neuroblastome de la cavité abdominale chez l'enfant, entraîne la formation de scellements palpables dans l'abdomen, une distension, une perte d'appétit, une constipation et une augmentation de la tension artérielle. Une tumeur qui comprime la moelle épinière ou une racine nerveuse peut entraîner un engourdissement et une faiblesse des membres, ainsi qu'une incapacité à se tenir debout, à ramper ou à marcher. Si les os sont touchés, des douleurs osseuses peuvent survenir.
En cas de tumeur de stade 3-4 dans la cavité abdominale avec atteinte des ganglions lymphatiques, les cellules tumorales peuvent pénétrer dans le parenchyme rénal, puis un neuroblastome étendu du rein chez l'enfant se développe, entraînant une perturbation de ses fonctions.
Étapes
- Le neuroblastome de stade 1 est une tumeur primaire localisée et isolée dans une zone du corps; les ganglions lymphatiques des deux côtés ne sont pas affectés.
- Neuroblastome de stade 2. Au stade 2A, la tumeur primitive est limitée à une zone, mais de grande taille; les ganglions lymphatiques bilatéraux ne sont pas atteints. Au stade 2B, les ganglions lymphatiques du côté du corps où se situe la tumeur sont positifs aux métastases.
- Neuroblastome stade 3: la tumeur primaire traverse la moelle épinière ou la ligne médiane du corps, des métastases unilatérales ou bilatérales sont retrouvées dans les ganglions lymphatiques.
- Neuroblastome de stade 4: la tumeur s'est propagée à des ganglions lymphatiques distants, à la moelle osseuse, aux os, au foie ou à d'autres organes. Le stade 4S est déterminé chez les enfants de moins d'un an présentant une tumeur primitive localisée, avec dissémination à la peau, au foie ou à la moelle osseuse.
Système international de classification des risques du neuroblastome (INRGSS)
L'INRGSS utilise les facteurs de risque définis par l'imagerie (IDRF), qui sont des facteurs observés lors des tests d'imagerie et qui peuvent signifier qu'une tumeur sera plus difficile à retirer.
L'INRGSS divise les neuroblastomes en 4 stades:
- L1: La tumeur ne s'est pas propagée depuis son point d'origine et n'a pas envahi les structures vitales. Elle est limitée à une partie du corps, comme le cou, le thorax ou l'abdomen.
- L2: La tumeur ne s'est pas propagée (métastase) loin de l'endroit où elle a commencé (par exemple, elle peut s'être développée du côté gauche de l'abdomen vers le côté gauche de la poitrine), mais elle présente au moins un IDRF.
- M: La tumeur a métastasé dans une partie éloignée du corps (sauf pour les tumeurs au stade SEP).
- SEP: Maladie métastatique chez les enfants de moins de 18 mois, dans laquelle le cancer s’est propagé uniquement à la peau, au foie et/ou à la moelle osseuse.
Complications et conséquences
Le neuroblastome se caractérise par des complications et des conséquences telles que:
- propagation (métastases) aux ganglions lymphatiques, à la moelle osseuse, au foie, à la peau et aux os;
- compression de la moelle épinière (qui peut causer de la douleur et conduire à une paralysie);
- développement d' un syndrome paranéoplasique (dû à l'action de certains produits chimiques sécrétés par la tumeur, ainsi que de l'antigène disialoganglioside GD2 exprimé par ses cellules), qui se manifeste par des mouvements oculaires involontaires rapides, une altération de la coordination, des crampes musculaires et de la diarrhée;
- rechutes après la fin du traitement primaire (comme le montre la pratique clinique, les neuroblastomes à haut risque présentent une rechute dans 50 % des cas).
Diagnostics neuroblastomes
Le diagnostic d’un neuroblastome suspecté chez un enfant nécessite un examen, des tests de laboratoire et une imagerie.
Des analyses de sang et d'urine sont effectuées pour détecter les catécholamines (noradrénaline et dopamine) et les acides homovanillique ou vanillylmandélique (formés lors du métabolisme de ces hormones); une analyse sanguine pour l'énolase neurospécifique, un test ELISA (enzyme-linked immunosorbent assay) du sérum sanguin et une analyse de moelle osseuse (dont un échantillon est prélevé par ponction-aspiration). Un test ADN est réalisé pour déterminer les mutations et une biopsie est réalisée pour un examen cytomorphologique du tissu tumoral.
Une fois les échantillons prélevés, ils sont envoyés au laboratoire où ils sont examinés au microscope par un pathologiste (médecin spécialisé dans l'identification des cellules cancéreuses). Des analyses spécifiques sont également souvent effectuées sur les échantillons afin de déterminer s'il s'agit d'un neuroblastome.
S’il s’agit d’un neuroblastome, des tests en laboratoire peuvent également aider à déterminer la vitesse à laquelle la tumeur pourrait se développer ou se propager, ainsi que les traitements qui pourraient être les plus efficaces.
Le diagnostic instrumental permet de visualiser le néoplasme à l'aide d'une échographie, d'une radiographie, d'une IRM ou d'une TDM, d'une TEP avec introduction de 18F-fluorodésoxyglucose ou d'une scintigraphie MIBG - scintigraphie à la métaiodobenzylguanidine. [ 1 ]
Diagnostic différentiel
Le diagnostic différentiel comprend le ganglioneurome bénin, le ganglioneuroblastome, le rhabdomyosarcome et le néphroblastome.
Qui contacter?
Traitement neuroblastomes
Dans le cas du neuroblastome, le traitement dépend du groupe à risque du patient (stade tumoral), de la localisation de la tumeur, des caractéristiques génomiques des cellules tumorales et de l'âge de l'enfant. Il peut inclure une surveillance, une intervention chirurgicale, une chimiothérapie, une radiothérapie, une immunothérapie et une greffe de cellules souches hématopoïétiques.
La chimiothérapie néoadjuvante ou adjuvante (pré- ou postopératoire) du neuroblastome chez l'enfant, comme toute chimiothérapie anticancéreuse, est administrée par cycles: le médicament est administré pendant plusieurs jours consécutifs, suivis d'une pause pour permettre à l'organisme de récupérer. Les cycles sont généralement répétés toutes les trois à quatre semaines.
Les médicaments suivants (et leurs combinaisons) sont utilisés: cyclophosphamide, cisplatine ou carboplatine, doxorubicine (adriamycine), vincristine, étoposide.
Les effets secondaires courants des médicaments de chimiothérapie comprennent la perte de cheveux, la perte d'appétit, la fatigue, les nausées et vomissements, les aphtes, la diarrhée ou la constipation. La chimiothérapie peut endommager la moelle osseuse et entraîner une diminution du nombre de cellules sanguines.
L'immunothérapie ciblée (ciblant l'antigène tumoral GD2) utilise des anticorps monoclonaux (AcM anti-GD2) du groupe des dinutuximab (Unituxin) et du naxitamab. Ils sont administrés par voie intraveineuse en perfusion prolongée, en association avec le facteur de stimulation des colonies de granulocytes et de macrophages (cytokine GM-CSF) et l'interleukine-2.
Les effets secondaires de ces médicaments comprennent des douleurs (souvent très intenses), une diminution de la pression artérielle, une augmentation du rythme cardiaque, un essoufflement (avec gonflement possible des voies respiratoires), une augmentation de la température, des nausées, des vomissements et de la diarrhée, ainsi que des modifications de la composition cellulaire et minérale du sang.
Pour réduire le risque de récidive du cancer après une chimiothérapie à haute dose et une transplantation de cellules souches, les enfants atteints d'un neuroblastome à haut risque sont traités avec des rétinoïdes systémiques, l'acide 13-cis-rétinoïque (isotrétinoïne). [ 2 ]
Traitement chirurgical du neuroblastome – ablation de la tumeur, par exemple, surrénalectomie ouverte ou résection laparoscopique du neuroblastome surrénalien; lymphectomie (ablation des ganglions lymphatiques affectés), etc. [ 3 ]
Pour le neuroblastome à haut risque, la radiothérapie peut être utilisée.[ 4 ]
La prévention
Compte tenu des causes du neuroblastome chez l'enfant, la seule mesure préventive pourrait être un conseil génétique lors de la planification d'une grossesse. Il convient toutefois de garder à l'esprit que cette tumeur n'est associée à des mutations héréditaires que dans 1 à 2 % des cas.
Prévoir
Le neuroblastome infantile a la capacité de régresser spontanément.
Marqueurs pronostiques
- Les tumeurs à haut risque, ainsi que le neuroblastome chez les enfants de tous âges et de tous stades (sauf le stade 4S) – avec une expression accrue du gène N-MYC et une amplification de l'oncogène N-Myc – ont un pronostic défavorable qui affecte l'espérance de vie.
- La présence de cellules tumorales dépourvues de certaines parties des chromosomes 1 ou 11 (délétions 1p ou 11q) entraîne un pronostic plus sombre. La présence d'une partie supplémentaire du chromosome 17 (gain 17q) est également associée à un pronostic plus sombre.
- Les cellules de neuroblastome contenant une grande quantité d’ADN ont un meilleur pronostic, en particulier chez les enfants de moins de 2 ans.
- Les neuroblastomes qui possèdent davantage de récepteurs de neurotrophine, en particulier le récepteur du facteur de croissance nerveuse TrkA, ont un meilleur pronostic.
Survie par groupe de risque du groupe d'oncologie infantile (COG)
- Groupe à faible risque: les enfants du groupe à faible risque ont un taux de survie à 5 ans supérieur à 95 %.
- Groupe à risque intermédiaire: Les enfants du groupe à risque intermédiaire ont un taux de survie à 5 ans de 90 % à 95 %.
- Groupe à haut risque: les enfants du groupe à haut risque ont un taux de survie à 5 ans d’environ 50 %.
Environ 15 % des décès par cancer chez l'enfant sont dus au neuroblastome. Les chances de survie à long terme pour cette tumeur maligne à haut risque ne dépassent pas 40 %. Le taux de survie global à cinq ans est de 67 à 74 %, 43 % dans la tranche d'âge de un à quatre ans et plus de 80 % pour un neuroblastome diagnostiqué au cours de la première année de vie.
Использованная литература