Expert médical de l'article

Nouvelles publications
Maladie de Charcot-Marie-Tooth.
Dernière revue: 12.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
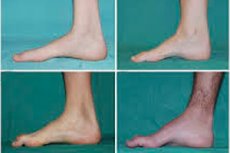
L'atrophie musculaire péronière, ou syndrome ou maladie de Charcot-Marie-Tooth, est un groupe de maladies héréditaires chroniques avec atteinte des nerfs périphériques.
Selon la CIM-10, dans la section sur les maladies du système nerveux, le code de cette maladie est G60.0 (neuropathie motrice et sensitive héréditaire). Elle figure également sur la liste des maladies orphelines.
Épidémiologie
Selon les statistiques cliniques, la prévalence de tous les types de maladie de Charcot-Marie-Tooth pour 100 000 habitants est de 19 cas (selon d'autres sources, un cas pour 2,5 à 10 000 habitants).
La CMT de type 1 représente environ les deux tiers des cas (un cas pour 5 000 à 7 000 habitants), et près de 70 % d'entre eux sont associés à une duplication du gène PMP22. Plus de 1,2 million de personnes dans le monde souffrent de ce type de maladie.
L’incidence du CMT de type 4 est estimée à 1 à 5 cas pour 10 000 enfants. [ 1 ]
Causes Maladie de Charcot-Marie-Tooth
Selon la classification des syndromes polyneuropathiques, l'atrophie musculaire péronière (fibulaire), l'amyotrophie neurale de Charcot-Marie-Tooth ou la maladie de Charcot-Marie-Tooth (en abrégé CMT) fait référence à des polyneuropathies motrices et sensorielles génétiquement déterminées. [ 2 ]
Autrement dit, les causes de son apparition sont des mutations génétiques. Selon la nature des anomalies génétiques, on distingue les principaux types de ce syndrome: démyélinisant et axonal. Le premier groupe comprend la maladie de Charcot-Marie-Tooth de type 1 (CMT1), due à la duplication du gène PMP22 sur le chromosome 17, codant pour la protéine transmembranaire de la myéline périphérique 22. Il en résulte une démyélinisation segmentaire de la gaine axonale (processus des cellules nerveuses) et une diminution de la vitesse de conduction du signal nerveux. De plus, des mutations peuvent affecter d'autres gènes.
La forme axonale est la maladie de Charcot-Marie-Tooth de type 2 (CMT2), qui affecte les axones eux-mêmes et est associée à des modifications pathologiques du gène MFN2 au locus 1p36.22, codant pour la protéine membranaire mitofusine-2, nécessaire à la fusion mitochondriale et à la formation de réseaux mitochondriaux fonctionnels à l'intérieur des cellules nerveuses périphériques. Il existe plus d'une douzaine de sous-types de CMT2 (avec des mutations de gènes spécifiques).
Il convient de noter qu'à l'heure actuelle, plus d'une centaine de gènes ont été identifiés dont les lésions, transmises héréditairement, sont à l'origine de divers sous-types de la maladie de Charcot-Marie-Tooth. Par exemple, des mutations du gène RAB7 conduisent à la CMT de type 2B; l'altération du gène SH3TC2 (codant une protéine membranaire des cellules de Schwann) provoque la CMT de type 4C, qui se manifeste dès l'enfance et se caractérise par une démyélinisation des neurones moteurs et sensoriels (il existe une douzaine et demie de formes de type 4 de cette maladie).
Un syndrome rare de type 3 (appelé syndrome de Dejerine-Sottas) commence à se développer dans la petite enfance et est causé par des mutations dans les gènes PMP22, MPZ, EGR2 et d'autres gènes.
Lorsque le CMT de type 5 survient entre 5 et 12 ans, on observe non seulement une neuropathie motrice (sous forme de paraparésie spastique des membres inférieurs), mais également des lésions des nerfs optiques et auditifs.
Une faiblesse musculaire et une atrophie optique (avec perte de vision) ainsi que des problèmes d'équilibre sont typiques de la CMT de type 6. Et dans la maladie de Charcot-Marie-Tooth de type 7, il n'y a pas seulement une neuropathie motrice-sensorielle, mais aussi une maladie rétinienne sous forme de rétinite pigmentaire.
Plus fréquente chez les hommes, la CMT liée à l'X ou maladie de Charcot-Marie-Tooth avec tétraparésie des membres (faiblesse des mouvements des bras et des jambes) est un type démyélinisant et on pense qu'elle résulte d'une mutation du gène GJB1 sur le bras long du chromosome X, qui code la connexine 32, une protéine transmembranaire des cellules de Schwann et des oligodendrocytes qui régule la transmission des signaux nerveux. [ 3 ]
Facteurs de risque
Le principal facteur de risque de CMT est une histoire familiale de la maladie, c'est-à-dire chez des proches parents.
Selon les généticiens, si les deux parents sont porteurs du gène autosomique récessif de la maladie de Charcot-Marie-Tooth, le risque d'avoir un enfant qui développera cette maladie est de 25 %. Le risque que l'enfant soit porteur de ce gène (sans présenter de symptômes) est estimé à 50 %.
En cas de transmission liée au chromosome X (lorsque le gène muté est présent sur le chromosome X de la femme), le risque que la mère transmette le gène à son fils est de 50 %. La maladie peut ne pas survenir à la naissance d'une fille, mais les fils (petits-enfants) de la fille peuvent hériter du gène défectueux et développer la maladie.
Pathogénèse
Dans tout type de maladie de Charcot-Marie-Tooth, sa pathogénèse est causée par une anomalie héréditaire des nerfs périphériques: moteurs (mouvement) et sensitifs (sensibles).
Si le type CMT est démyélinisant, la destruction ou le défaut de la gaine de myéline qui protège les axones des nerfs périphériques entraîne un ralentissement de la transmission des impulsions nerveuses dans le système nerveux périphérique - entre le cerveau, les muscles et les organes sensoriels.
Dans le type axonal de la maladie, les axones eux-mêmes sont affectés, ce qui affecte négativement la force des signaux nerveux, qui est insuffisante pour une stimulation complète des muscles et des organes sensoriels.
Lire aussi:
Comment le syndrome de Charcot-Marie-Tooth se transmet-il? Les gènes défectueux peuvent être transmis selon un mode autosomique dominant ou autosomique récessif.
Le type le plus courant, la transmission autosomique dominante, survient lorsqu'il existe une copie du gène muté (portée par l'un des parents). La probabilité de transmettre la CMT à chaque enfant né est estimée à 50 %. [ 4 ]
Dans le cas d’une transmission autosomique récessive, deux copies du gène défectueux (une de chaque parent ne présentant aucun signe de la maladie) sont nécessaires pour que la maladie se développe.
Dans 40 à 50 % des cas, on observe une démyélinisation héréditaire autosomique dominante, c'est-à-dire une CMT de type 1; dans 12 à 26 % des cas, une CMT axonale, c'est-à-dire de type 2. Et dans 10 à 15 % des cas, on observe une hérédité liée à l'X. [ 5 ]
Symptômes Maladie de Charcot-Marie-Tooth
Généralement, les premiers signes de cette maladie apparaissent dès l'enfance et l'adolescence et se développent progressivement tout au long de la vie, bien que le syndrome puisse se manifester plus tard. La combinaison des symptômes est variable, et la vitesse de progression de la maladie, ainsi que sa gravité, sont impossibles à prévoir.
Les symptômes typiques du stade initial comprennent une fatigue générale accrue; une diminution du tonus (faiblesse) des muscles des pieds, des chevilles et des tibias; et une perte de réflexes. Cela rend les mouvements du pied difficiles et entraîne une dysbasie (troubles de la marche) se traduisant par une élévation plus importante des jambes, souvent accompagnée de trébuchements et de chutes fréquents. Les signes de la maladie de Charcot-Marie-Tooth chez le jeune enfant peuvent inclure une maladresse prononcée et des difficultés de marche inhabituelles pour l'âge, associées à un pied tombant bilatéral. Des déformations du pied sont également caractéristiques: voûte plantaire haute (pied creux) ou pieds plats prononcés, orteils courbés (en marteau).
En cas de marche sur la pointe des pieds dans un contexte d'hypotonie musculaire, un neurologue peut suspecter que l'enfant est atteint de CMT de type 4, dans lequel les enfants peuvent être incapables de marcher à l'adolescence.
À mesure que la maladie progresse, l'atrophie et la faiblesse musculaires s'étendent aux membres supérieurs, rendant difficile la motricité fine et les tâches manuelles courantes. Une diminution des sensations tactiles et de la perception du chaud et du froid, ainsi qu'un engourdissement des pieds et des mains, indiquent une atteinte des axones des nerfs sensoriels.
Dans la maladie de Charcot-Marie-Tooth de types 3 et 6 qui se manifeste dans l'enfance, on observe une ataxie sensorielle (altération de la coordination des mouvements et de l'équilibre), des contractions musculaires et des tremblements, des lésions du nerf facial, une atrophie du nerf optique avec nystagmus et une perte auditive.
Dans les stades ultérieurs, des tremblements incontrôlables et des crampes musculaires fréquentes peuvent survenir; des problèmes de mouvement peuvent entraîner le développement de douleurs: musculaires, articulaires, neuropathiques.
Complications et conséquences
La maladie de Charcot-Marie-Tooth peut avoir des complications et des conséquences telles que:
- entorses et fractures plus fréquentes;
- contractures associées au raccourcissement des muscles et des tendons périarticulaires;
- scoliose (courbure de la colonne vertébrale);
- problèmes respiratoires – lorsque les fibres nerveuses qui innervent les muscles du diaphragme sont endommagées:
- perte de la capacité de se déplacer de façon autonome.
Diagnostics Maladie de Charcot-Marie-Tooth
Le diagnostic comprend l’examen clinique, l’anamnèse (y compris les antécédents familiaux), l’examen neurologique et systémique.
Des tests sont effectués pour vérifier l'amplitude des mouvements, la sensibilité et les réflexes tendineux. La conduction nerveuse peut être évaluée à l'aide d'instruments diagnostiques tels que l'électromyographie ou l'électroneuromyographie. Une échographie ou une IRM peuvent également être nécessaires. [ 6 ]
Les tests génétiques ou ADN permettant de détecter les mutations génétiques les plus courantes responsables de la CMT dans un échantillon sanguin sont limités, car les tests ADN ne sont pas actuellement disponibles pour tous les types de maladie. Pour plus d'informations, consultez la section « Tests génétiques ».
Dans certains cas, une biopsie du nerf périphérique (généralement le nerf sural) est réalisée.
Diagnostic différentiel
Le diagnostic différentiel inclut d'autres neuropathies périphériques, la dystrophie musculaire de Duchenne, les syndromes myélopathiques et myasthéniques, la neuropathie diabétique, les myélopathies dans la sclérose latérale multiple et amyotrophique, le syndrome de Guillain-Barré, un traumatisme du nerf péronier et son atrophie (y compris lorsqu'il est pincé entre les disques lombaires de la colonne vertébrale), des lésions du cervelet ou du thalamus, ainsi que les effets secondaires de la chimiothérapie (pendant le traitement par des cytostatiques tels que la vincristine ou le paclitaxel). [ 7 ]
Qui contacter?
Traitement Maladie de Charcot-Marie-Tooth
Aujourd'hui, le traitement de cette maladie héréditaire comprend la rééducation par l'exercice (visant à renforcer et à étirer les muscles); l'ergothérapie (qui aide les patients souffrant de faiblesse musculaire des mains); et l'utilisation d'appareils orthopédiques pour faciliter la marche. Si nécessaire, des analgésiques ou des anticonvulsivants sont prescrits. [ 8 ]
En cas de pieds plats sévères, une ostéotomie peut être réalisée, et en cas de déformation du talon, une correction chirurgicale est indiquée – l’arthrodèse. [ 9 ]
Des recherches sont en cours sur la composante génétique de la maladie et ses traitements. L'utilisation de cellules souches, de certaines hormones, de lécithine ou d'acide ascorbique n'a pas encore donné de résultats positifs.
Mais grâce à des recherches récentes, une nouvelle approche pourrait bien émerger dans le traitement de la maladie de Charcot-Marie-Tooth dans un avenir proche. Ainsi, depuis 2014, la société française Pharnext développe, et depuis mi-2019, des essais cliniques sont en cours pour le médicament PXT3003 pour le traitement de la CMT de type 1 chez l'adulte. Ce médicament supprime l'expression accrue du gène PMP22, améliore la myélinisation des nerfs périphériques et soulage les symptômes neuromusculaires.
Des spécialistes de la société médicale Sarepta Therapeutics (États-Unis) travaillent à la création d'une thérapie génique pour la maladie de Charcot-Marie-Tooth de type 1. Cette thérapie utilisera un virus adéno-associé (AAV) inoffensif du genre Dependovirus, doté d'un génome d'ADN monocaténaire linéaire, qui transférera dans l'organisme le gène NTF3, codant pour la protéine neurotrophine-3 (NT-3), nécessaire au fonctionnement des cellules nerveuses de Schwann.
Helixmith commencera les essais cliniques de la thérapie génique Engensis (VM202) développée en Corée du Sud d'ici la fin de 2020 pour traiter les symptômes musculaires de la CMT de type 1. [ 10 ]
La prévention
La prévention de la CMT peut passer par un conseil génétique aux futurs parents, notamment si un membre du couple présente des antécédents familiaux de la maladie. Cependant, des cas de mutations génétiques ponctuelles de novo ont été identifiés, c'est-à-dire en l'absence de la maladie dans les antécédents familiaux.
Pendant la grossesse, la probabilité de maladie de Charcot-Marie-Tooth chez le futur enfant peut être vérifiée par une biopsie des villosités choriales (de 10 à 13 semaines de gestation), ainsi qu'une analyse du liquide amniotique (à 15-18 semaines).
Prévoir
En général, le pronostic des différents types de maladie de Charcot-Marie-Tooth dépend de la gravité clinique, mais dans tous les cas, la maladie évolue lentement. De nombreux patients présentent des handicaps, sans pour autant réduire leur espérance de vie.