Expert médical de l'article

Nouvelles publications
Néphrite héréditaire (syndrome d'Alport) chez les enfants
Dernière revue: 23.04.2024

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Hereditary néphrite (syndrome de Alport) - génétiquement déterminée glomérulopathie héréditaire non-immune présentant une hématurie (parfois protéinurie), la diminution progressive de la fonction rénale dans le développement de l'insuffisance rénale chronique est souvent associée à une surdité de perception et aux malvoyants.
Pour la première fois la maladie a été décrite en 1902 par LG Guthrie, qui a observé une famille dans plusieurs générations dont une hématurie a été observée. En 1915, des membres de la même famille AFHurst ont décrit le développement de l'urémie. En 1927, A Alport a identifié la surdité de plusieurs membres de sa famille atteints d'hématurie et, dans les années 50 du siècle dernier, des lésions oculaires ont été décrites dans une telle maladie. En 1972, chez des patients atteints d'hématurie héréditaire, examinant morphologiquement le tissu rénal, Hinglais et al. Révélé une expansion inégale et la délamination des membranes basales glomérulaires. En 1985, la base génétique de la néphrite héréditaire - une mutation dans le gène du collagène de type IV (Fiengold et al., 1985) a été identifiée.
L'étude de la nature génétique de la maladie a permis de conclure que les différences dans les manifestations phénotypiques de la néphrite héréditaire (avec ou sans perte auditive) sont dues au degré d'expression du gène mutant. Ainsi, à l'heure actuelle, tous les variants cliniques sont considérés comme des manifestations d'une maladie et le terme «néphrite héréditaire» est synonyme du terme «syndrome d'Alport».
Selon des études épidémiologiques, la néphrite héréditaire se produit à une fréquence de 17 pour 100 000 enfants.
Causes du syndrome d'Alport
La base génétique de la maladie est une mutation du gène a-5 de la chaîne collagène de type IV. Ce type est universel pour les membranes basales du rein, la cochlée, la capsule du cristallin, la rétine et la cornée de l'œil, ce qui est prouvé dans des études utilisant des anticorps monoclonaux contre cette fraction de collagène. Récemment, ils indiquent la possibilité d'utiliser des sondes d'ADN pour le diagnostic prénatal de la néphrite héréditaire.
L'importance de tester tous les membres de la famille en utilisant des sondes d'ADN pour identifier les porteurs du gène mutant est soulignée, ce qui est d'une grande importance dans la conduite de conseil génétique génétique des familles atteintes de cette maladie. Cependant, jusqu'à 20% des familles n'ont pas de famille avec une maladie rénale, ce qui suggère une forte incidence de mutations spontanées dans le gène anormal. La majorité des patients atteints de néphrite héréditaire dans les familles ont des personnes atteintes d'une maladie rénale, d'une perte auditive et d'une pathologie de la vision; les mariages apparentés entre personnes ayant un ou plusieurs ancêtres, puisque le mariage d'individus apparentés augmente la probabilité d'obtenir les mêmes gènes chez les deux parents. Autosomique dominante et autosomique récessive et dominante, liée au chromosome X de la voie de transmission sont établies.
Les enfants sont plus susceptibles de distinguer trois variantes de la néphrite héréditaire: le syndrome d'Alport, la néphrite héréditaire sans perte d'audition et l'hématurie bénigne de la famille.
Alport Syndrome - néphrite héréditaire avec des troubles de l'audition. La base est un défaut combiné dans la structure du collagène de la membrane basale des glomérules des reins, les structures de l'oreille et des yeux. Le gène du syndrome d'Alport classique est situé au locus 21-22 q du bras long du chromosome X. Dans la plupart des cas, il est hérité par le type dominant lié au chromosome X. À cet égard, chez les hommes, le syndrome d'Alport est plus difficile, car chez les femmes, la fonction du gène mutant est compensée par un allèle sain du second chromosome intact.
Les bases génétiques du développement de la néphrite héréditaire sont des mutations dans les gènes des chaînes alpha du collagène de type IV. Il est connu que six chaînes de collagène de type IV G: les gènes A5 et A6 chaînes (Sol4A5 et Sol4A5) sont situées sur le bras long du chromosome X dans la zone 21-22q; les gènes des chaînes a3 et a4 (Co4A3 et Co4A4) - sur le chromosome 2-ème; les gènes des chaînes a1 et a2 (Co4A1 et Co4A2) - sur le 13ème chromosome.
Dans la plupart des cas (80-85%), un type d'hérédité liée à l'X est associé à une lésion du gène Co4A5 due à une délétion, à des mutations ponctuelles ou à des troubles de l'épissage. Actuellement, plus de 200 mutations du gène Kol4A5, responsable de la violation de la synthèse des chaînes a5 du collagène de type IV, sont retrouvées. Dans ce type d'hérédité, la maladie se manifeste chez les enfants des deux sexes, mais chez les garçons, elle est plus difficile.
Des mutations dans les locus des gènes Co4A3 et Co4A4, responsables de la synthèse des chaînes a3 et a4 du collagène de type IV, sont héritées autosomiquement. Selon la recherche, le type de transmission autosomique dominant est observé dans 16% des cas de néphrite héréditaire, autosomique récessive - chez 6% des patients. Il y a environ 10 mutations des gènes de Co4A3 et Co4A4.
Le résultat de mutations est une violation des processus d'assemblage du collagène de type IV, entraînant une perturbation de sa structure. Le collagène de type IV est l'un des composants principaux de la membrane basale glomérulaire, de l'appareil cochléaire et du cristallin, dont la pathologie sera révélée en clinique de néphrite héréditaire.
Le collagène de type IV, une partie de la membrane basale glomérulaire, est essentiellement constitué de deux chaînes A1 (IV) et une chaîne a2 (IV), et contient également a3, a4, a5 chaîne. Le plus souvent, lorsque la mutation Sol4A5 héritage lié à l'X accompagné d'un manque a3, A4- et a6 chaînes A5 de collagène de type IV dans la structure, et le nombre de chaînes O1 et a2 dans l'augmentation de la membrane basale glomérulaire. Le mécanisme de ce phénomène n'est pas clair, on suppose que la cause est les changements post-transcriptionnels dans l'ARNm.
Le manque a3, A4-, et des chaînes a5 dans le type de structure membrane basale collagène IV des résultats glomérules dans l'amincissement et la fragilité des stades précoces du syndrome d'Alport qui se manifeste cliniquement plupart hématurie (parfois hématurie ou protéinurie seulement protéinurie), la perte et lenticône auditive. En outre la progression de la maladie conduit à un épaississement et une perturbation de la perméabilité de la membrane basale dans les derniers stades de la maladie, avec la croissance de ces types de collagène V et VI, qui se manifeste dans l'augmentation de la protéinurie et la réduction de la fonction rénale.
La nature de la mutation sous-jacente à la néphrite héréditaire détermine en grande partie sa manifestation phénotypique. Lorsque des deletions du chromosome X avec mutation simultanée et gènes Sol4A6 Sol4A5 responsables de la synthèse de A5 et A6 chaînes de collagène de type IV, combinés avec l'oesophage du syndrome d'Alport de léiomyomatose et les organes génitaux. Selon des études avec des mutations du gène Sol4A5 associées à une suppression sont marqués grande sévérité du processus pathologique, une combinaison avec une lésion rénale des manifestations extra-rénales et le développement précoce de l'insuffisance rénale chronique, une mutation par rapport stochechnoy de ce gène.
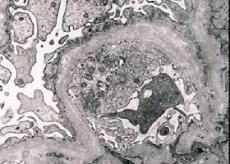
Morphologiquement, la microscopie électronique révèle l'amincissement et la délamination des membranes basales glomérulaires (en particulier lamina densa) et la présence de granules électroniquement denses. La lésion du glomérule peut être non uniforme chez le même patient, de la lésion focale minimale du mésangium à la glomérulosclérose. La glomérulite dans le syndrome d'Alport est toujours immuno-négative, ce qui la distingue de la glomérulonéphrite. Caractéristique sont le développement de l'atrophie canalaire, l'infiltration lymphohistiocyte, la présence de "cellules de mousse" avec des inclusions de lipides - lipofagi. Avec la progression de la maladie, un épaississement et une destruction marquée des membranes glomérulaires basales sont révélés.
Certains changements dans l'état du système immunitaire sont révélés. Les patients atteints de néphrite héréditaire diminue le taux d'Ig A, et une tendance à une augmentation de la concentration sanguine d'IgM, IgG niveau peut être augmenté dans les premiers stades de la maladie et le déclin dans les étapes ultérieures. Peut-être une augmentation de la concentration d'IgM et G est une sorte de réponse compensatoire en réponse à un déficit en IgA.
L'activité fonctionnelle du système des lymphocytes T est réduite; il y a une diminution sélective des lymphocytes B responsables de la synthèse de Ig A, le lien phagocytaire de l'immunité est violé, principalement en raison de la violation de la chimiotaxie et de la digestion intracellulaire dans les neutrophiles
Dans l'étude de la biopsie rénale chez les patients atteints du syndrome d'Alport par microscopie électronique, les changements ultrastructuraux observés membrane basale glomérulaire: amincissement, et des motifs de séparation violation des membranes basales glomérulaires avec le changement de son épaisseur et les contours irréguliers. Dans les premiers stades de la néphrite héréditaire, le défaut détermine l'amincissement et la fragilité des membranes basales glomérulaires.
L'amincissement des membranes glomérulaires est un signe plus favorable et plus fréquent chez les filles. Une caractéristique microscopique électronique plus constante dans la néphrite héréditaire est le clivage de la membrane basale, et la sévérité de sa destruction est en corrélation avec la gravité du processus.

Les symptômes du syndrome d'Alport chez les enfants
Les premiers symptômes du syndrome d'Alport sous forme de syndrome urinaire isolé sont plus souvent détectés chez les enfants des trois premières années de vie. Dans la plupart des cas, la maladie est détectée par accident. Le syndrome urinaire est révélé lors de l'examen préventif de l'enfant, avant d'entrer dans l'institution de l'enfant ou au cours de l'ARVI. En cas d'apparition de pathologie dans l'urine lors d'ARVI. Dans la néphrite héréditaire, contrairement à la glomérulonéphrite acquise, il n'y a pas de période de latence.
Au stade initial de la maladie, le bien-être de l'enfant souffre peu, la caractéristique étant la persistance et la persistance du syndrome urinaire. L'un des principaux signes est une hématurie de divers degrés, observée dans 100% des cas. L'augmentation du degré d'hématurie est notée pendant ou après les infections des voies respiratoires, l'effort physique ou après des vaccinations préventives. Protéinurie dans la plupart des cas ne dépasse pas 1 g / jour, au début de la maladie peut être instable, à mesure que le processus progresse la protéinurie augmente. Périodiquement, les sédiments urinaires peuvent avoir une leucocyturie avec une prédominance de lymphocytes, ce qui est associé au développement de changements interstitiels.
Plus tard, il y a une violation des fonctions partielles des reins, aggravation de l'état général du patient: intoxication, faiblesse musculaire, hypotension artérielle, souvent déficience auditive (en particulier chez les garçons), parfois une vision altérée. L'intoxication se manifeste par la pâleur, la fatigue, les maux de tête. Au stade initial de la maladie, la perte d'audition dans la plupart des cas n'est détectée que par audiographie. La perte d'audition dans le syndrome d'Alport peut survenir à différentes périodes de l'enfance, mais le plus souvent la perte d'audition est diagnostiquée à l'âge de 6-10 ans. La surdité chez les enfants commence à des fréquences élevées, atteignant un degré considérable de conduction aérienne et osseuse, passant de la surdité sonore à la surdité sonore. La perte auditive peut être l'un des premiers symptômes de la maladie et peut précéder le syndrome urinaire.
Dans 20% des cas, les patients atteints du syndrome d'Alport ont des changements dans les yeux. Les anomalies les plus communes de la lentille: sphérofokiya, lentikonus antérieur, postérieur ou mixte, une variété de cataractes. Dans les familles atteintes du syndrome d'Alport, il existe une incidence significative de myopie. Un certain nombre de chercheurs constamment dans ces familles notent des changements périmaculaires bilatéraux sous la forme de granulations blanchâtres ou jaunâtres brillantes dans la zone du corps jaune. Ils considèrent ce symptôme comme un symptôme constant, qui a une grande valeur diagnostique dans le syndrome d'Alport. C. S. Chugh et al. (1993) pour l'étude ont révélé des patients ophtalmologiques syndrome Alport diminution de l'acuité visuelle dans 66,7% des cas, l'lenticône avant - 37,8%, les taches sur la rétine - dans la cataracte, de 22,2% - 20%, kératocône - 6 , 7%.
Chez certains enfants atteints de néphrite héréditaire, en particulier dans la formation de l'insuffisance rénale, un retard important dans le développement physique est noté. Comme la progression de l'insuffisance rénale développe une hypertension. Chez les enfants, il est plus souvent détecté à l'adolescence et dans les groupes d'âge plus âgés.
Caractéristique est la présence chez les patients atteints de néphrite héréditaire de divers (plus de 5-7) stigmates de la dysembryogenèse du tissu conjonctif. Parmi le tissu conjonctif de la stigmatisation chez les patients atteints hypertelorism l'œil le plus fréquent, le palais haut, malocclusion, forme anormale des oreilles, la courbure du petit doigt sur ses mains, « l'écart de sandalevidnaya » sur les pieds. Car la néphrite héréditaire est caractérisée par le même type de stigmatisation de la dizembryogenèse au sein de la famille, ainsi que par la fréquence élevée de leur propagation parmi les parents des probants, à travers lesquels la maladie est transmise.
Dans les premiers stades de la maladie a révélé une réduction isolée de la fonction rénale partielle: transport des acides aminés, des électrolytes, des fonctions de concentration acidogenesis, d'autres modifications sont état de fonctionnement à la fois du néphron proximale et distale et ont le caractère de troubles partiels combinés. La réduction de la filtration glomérulaire se produit plus tard, plus souvent à l'adolescence. À mesure que la néphrite héréditaire progresse, l'anémie se développe.
Ainsi, pour une néphrite héréditaire mise en scène caractérisé de la maladie: première phase latente ou symptômes cliniques cachés se manifestant par un minimum de modifications du syndrome de la vessie se produit alors processus de décompensation progressive avec une réduction de la fonction rénale avec des symptômes cliniques manifestes (intoxication, asthénie, des retards de développement, anemizatsiya). Les symptômes cliniques apparaissent généralement indépendamment de la stratification de la réaction inflammatoire.
Néphrite héréditaire peut se manifester dans différentes périodes d'âge, qui dépend de l'action du gène, qui jusqu'à un certain temps est dans un état refoulé.
Classification
Il existe trois variantes de la néphrite héréditaire
- I variante - se manifeste cliniquement par une néphrite avec hématurie, perte d'audition et des lésions oculaires. L'évolution de la néphrite est progressive avec le développement de la CRF. Le type d'hérédité est dominant, lié au chromosome X. Morphologiquement, il y a une perturbation de la structure de la membrane basale, son amincissement et son clivage.
- II variante - se manifeste cliniquement par une néphrite avec hématurie sans perte d'audition. L'évolution de la néphrite est progressive avec le développement de l'insuffisance rénale chronique. Le type d'hérédité est dominant, lié au chromosome X. Morphologiquement, l'amincissement de la membrane basale des capillaires glomérulaires (en particulier laminadensa) est révélé.
- Option III - hématurie bénigne de la famille. Le cours est favorable, l'insuffisance rénale chronique ne se développe pas. Le type de transmission est autosomique dominant ou autosomique récessif. Dans le type de transmission autosomique récessif, les femmes ont un cours plus sévère de la maladie.
Diagnostic du syndrome d'Alport
Les critères suivants sont proposés:
- la présence dans chaque famille d'au moins deux patients atteints de néphropathie;
- hématurie comme le symptôme principal de la néphropathie dans le proband;
- au moins un membre de la famille a une perte auditive;
- le développement de l'insuffisance rénale chronique chez un parent et plus.
Dans le diagnostic d'une variété de maladies héréditaires et congénitales une place importante appartient à une approche intégrée de l'inspection et surtout en faisant attention aux données obtenues dans la préparation du pedigree de l'enfant. Syndrome de diagnostic Alport jugé valable dans les cas où le patient 3 sur 4 caractéristiques typiques: la présence dans la hématurie familiale et l'insuffisance rénale chronique, la présence de la perte auditive neurosensorielle du patient, les pathologies de la détection des signes de biopsie de caractérisation au microscope électronique clivage membrane basale glomérulaire avec un changement de son épaisseur et des contours inégaux.
L'examen du patient devrait inclure des méthodes d'investigation génétique clinique; étude dirigée de l'anamnèse de la maladie; examen général du patient en tenant compte des critères de diagnostic. Au stade de la compensation, on ne peut attraper la pathologie qu'en se concentrant sur des syndromes tels que la présence de complication héréditaire, l'hypotension, la stigmatisation multiple de la dysembryogenèse, les modifications du syndrome urinaire. Au stade de la décompensation, il peut y avoir l'apparition de symptômes œstrogéniques, tels qu'une intoxication sévère, une asthénie, un retard de développement physique, une anémie, se manifestant et amplifiés avec une diminution progressive de la fonction rénale. Chez la plupart des patients présentant une diminution de la fonction rénale, on observe une diminution de la fonction de l'acido- et de l'aminogénèse; chez 50% des patients, on observe une diminution significative de la fonction sécrétoire des reins; limiter l'amplitude des fluctuations de la densité optique de l'urine; une violation du rythme de filtration, puis une diminution de la filtration glomérulaire. Le stade de l'insuffisance rénale chronique est diagnostiqué chez les patients ayant un taux d'urée de 3 à 6 mois et plus dans le sérum sanguin (plus de 0,35 g / l), une diminution de la filtration glomérulaire à 25% de la norme.
Le diagnostic différentiel de la néphrite héréditaire doit être effectuée principalement avec la forme acquise hematuric glomérulonéphrite. A gagné glomérulonéphrite de plus en plus aiguë période débutant 2-3 semaines après une infection antérieure, les caractéristiques extrarénales, y compris l'hypertension avec les premiers jours (en néphrite héréditaire, à l'inverse, l'hypotension), une diminution de la filtration glomérulaire au début, aucune violation des fonctions tubulaires partielles, alors que comme avec héréditaire ils sont présents. Glomérulonéphrite acquise se produit avec hématurie plus sévère et protéinurie, avec ESR accrue. Valeur diagnostique sont typiques des changements dans la membrane basale glomérulaire, la néphrite héréditaire caractéristique.
Le diagnostic différentiel de la néphropathie dysmétabolique réalisée avec l'insuffisance rénale chronique dans la famille identifié une maladie rénale cliniquement Monotype, et peut aller de néphropathies pyélonéphrite à urolithiase. Les enfants ont souvent des plaintes de douleurs abdominales et périodiquement avec la miction, dans les sédiments urinaires - oxalate.
Si vous soupçonnez qu'un patient néphrite héréditaire doit être envoyé pour clarifier le diagnostic dans un service spécialisé en néphrologie.
Qu'est-ce qu'il faut examiner?
Comment examiner?
Quels tests sont nécessaires?
Qui contacter?
Traitement du syndrome d'Alport
Dans le régime prévoit une restriction de l'effort physique important, rester dans l'air frais. Le régime est de haute qualité, avec une teneur suffisante en protéines, graisses et glucides de haute qualité, en tenant compte de la fonction des reins. L'identification et la réhabilitation des foyers chroniques d'infection revêtent une grande importance. De médicaments, ATP, cocarboxylase, pyridoxine (jusqu'à 50 mg / jour), le chlorure de carnitine sont utilisés. Les cours ont lieu 2-3 fois par an. Lorsque l'hématurie est prescrite phytothérapie - ortie, ortie, framboise, achillée millefeuille.
Dans la littérature étrangère et domestique, il existe des rapports de traitement avec la prednisolone et l'utilisation de cytostatiques. Cependant, l'effet est difficile à juger.
Dans l'insuffisance rénale chronique, l'hémodialyse et la transplantation rénale sont utilisées.
Il n'existe pas de méthode de traitement spécifique (pathogénique efficace) de la néphrite héréditaire. Toutes les mesures médicales visent à prévenir et à ralentir la réduction des fonctions rénales.
Le régime doit être équilibré et riche en calories, en tenant compte de l'état fonctionnel des reins. En l'absence de violations de l'état fonctionnel dans la nutrition de l'enfant devrait être un contenu suffisant de protéines, de graisses et de glucides. En présence de signes de dysfonctionnement rénal, la quantité de protéines, de glucides et de phosphore devrait être limitée, ce qui retarde le développement de l'insuffisance rénale chronique.
Le stress physique devrait être limité, les enfants devraient s'abstenir de faire du sport.
Éviter le contact avec les patients infectieux, réduire le risque de développer des infections respiratoires aiguës. Il est nécessaire de désinfecter les foyers d'infection chronique. Les vaccinations préventives pour les enfants atteints de néphrite héréditaire ne sont pas réalisées, la vaccination n'est possible que selon les indications épidémiologiques.
La thérapie hormonale et immunosuppressive dans la néphrite héréditaire est inefficace. Il existe des indications d'un certain effet positif (réduction de la protéinurie et ralentissement de la progression de la maladie) avec l'utilisation à long terme des inhibiteurs de la cyclosporine A et de l'ECA pendant de nombreuses années.
Dans le traitement des patients utilisant des médicaments qui améliorent le métabolisme:
- pyridoxine - 2-3 mg / kg / jour en 3 doses fractionnées pendant 4 semaines;
- kokarboksilaza - 50 mg par voie intramusculaire tous les deux jours, seulement 10-15 injections;
- ATP - 1 ml par voie intramusculaire tous les deux jours, 10-15 injections;
- Vitamine A - 1000 U / an / jour en 1 réception pendant 2 semaines;
- vitamine E - 1 mg / kg / jour dans 1 réception pendant 2 semaines.
Une telle thérapie améliore l'état général des patients, réduit le dysfonctionnement tubulaire, et est administrée 3 fois par an.
Comme un immunomodulateur peut être utilisé lévamisole - 2 mg / kg / jour 2-3 fois par semaine avec des interruptions entre les doses de 3-4 jours.
Pour les chercheurs, l'oxygénation hyperbare a un effet positif sur la sévérité de l'hématurie et de la dysfonction rénale.
La méthode la plus efficace de traiter la néphrite héréditaire est la transplantation de rein en temps opportun. Il n'y a pas de récurrence de la maladie dans la greffe, dans un faible pourcentage de cas (environ 5%), le développement de néphrite dans le rein greffé associé à des antigènes de la membrane basale glomérulaire est possible.
Un domaine prometteur est le diagnostic prénatal et la thérapie par génie génétique. Des expériences sur des animaux montrent une grande efficacité du transfert de gènes normaux responsables de la synthèse de chaînes a de collagène de type IV dans le tissu rénal, après quoi une synthèse de structures normales de collagène est notée.
Prévision
Le pronostic de la néphrite héréditaire est toujours sérieux.
Les critères pronostiques défavorables pour l'écoulement de la néphrite héréditaire sont:
- sexe masculin;
- le développement précoce de l'insuffisance rénale chronique chez les membres de la famille;
- protéinurie (plus de 1 g / jour);
- épaississement des membranes basales glomérulaires selon la microscopie;
- névrite du nerf auditif;
- délétion dans le gène Co4A5.
Le pronostic de l'hématurie bénigne de la famille est plus favorable.
Использованная литература