Expert médical de l'article

Nouvelles publications
Syndrome de Martin-Bell
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Le syndrome de Martin-Bell a été décrit en 1943 par des médecins, qui lui ont donné son nom. Il s'agit d'une maladie génétique caractérisée par un retard mental. En 1969, des modifications du chromosome X (fragilité du bras distal), caractéristiques de cette maladie, ont été identifiées. En 1991, des scientifiques ont découvert le gène responsable du développement de cette maladie. Cette maladie est également appelée « syndrome de l'X fragile ». Les garçons comme les filles sont sensibles à la maladie, mais les garçons sont plus souvent touchés (trois fois plus).
Épidémiologie
Le syndrome de Martin-Bell est une maladie assez fréquente: 0,3 à 1 homme sur 1 000 en souffre et 0,2 à 0,6 femme sur 1 000. De plus, les enfants atteints du syndrome de Martin-Bell naissent avec la même fréquence sur tous les continents. De toute évidence, la nationalité, la couleur de peau, la forme des yeux, les conditions de vie et le bien-être des personnes concernées n’ont pas d’influence sur l’apparition de la maladie. Sa fréquence n’est comparable qu’à celle du syndrome de Down (1 cas sur 600 à 800 nouveau-nés). Un cinquième des hommes porteurs du gène altéré sont en bonne santé, ne présentent aucune anomalie clinique ou génétique; les autres présentent des signes de retard mental, de formes légères à sévères. Parmi les femmes porteuses, un peu plus d’un tiers sont malades.
Le syndrome de l'X fragile touche environ 1 homme sur 2 500 à 4 000 et 1 femme sur 7 000 à 8 000. La prévalence des porteurs est estimée à 1 sur 130 à 250 chez les femmes et à 1 sur 250 à 800 chez les hommes.
Causes Syndrome de Martin-Bell
Le syndrome de Martin-Bell se développe suite à l'arrêt complet ou partiel de la production d'une protéine spécifique par l'organisme. Cela est dû à l'absence de réponse du gène FMR1, localisé sur le chromosome X. La mutation résulte de la restructuration du gène à partir de variants structurels instables des états géniques (allèles), et non dès le début. La maladie se transmet uniquement par voie masculine, et l'homme n'est pas nécessairement malade. Les porteurs mâles transmettent le gène à leurs filles sous une forme inchangée, de sorte que leur retard mental n'est pas évident. Lors d'une transmission ultérieure du gène de la mère à ses enfants, le gène mute et tous les signes caractéristiques de cette maladie apparaissent.
Pathogénèse
La pathogénèse du syndrome de Martin-Bell repose sur des mutations de l'appareil génétique qui bloquent la production de la protéine FMR, une protéine essentielle à l'organisme, notamment dans les neurones, et présente dans divers tissus. Les recherches montrent que les protéines FMR sont directement impliquées dans les processus de régulation des traductions qui se produisent dans le tissu cérébral. L'absence de cette protéine ou sa production limitée par l'organisme entraîne un retard mental.
Dans la pathogenèse de la maladie, l’hyperméthylation des gènes est considérée comme un trouble clé, mais il n’a pas encore été possible d’identifier définitivement le mécanisme de développement de ce trouble.
Parallèlement, une hétérogénéité locus de la pathologie a été découverte, associée à un polyallélisme et à un polylocus. La présence de variants alléliques responsables du développement de la maladie a été déterminée, causés par des mutations ponctuelles et la destruction du gène de type FMRL.
Les patients présentent également deux triplets fragiles sensibles à l'acide folique, situés à 300 kb, ainsi que 1,5 à 2 millions de pb provenant du triplet fragile contenant le gène FMR1. Le mécanisme des mutations des gènes FRAXE et FRAXF (identifiées dans les triplets fragiles mentionnés ci-dessus) est lié à celui des troubles du syndrome de Martin Bell. Ce mécanisme est dû à la propagation des répétitions GCC et CGG, qui entraînent la méthylation des îlots CpG. Outre la forme classique de la pathologie, il existe également deux types rares qui se distinguent par l'expansion des répétitions de trinucléotides (en méiose mâle et femelle).
Il a été constaté que dans la forme classique du syndrome, le patient est dépourvu d'une protéine nucléocytoplasmique spécifique de type FMR1, qui assure la liaison de divers ARNm. De plus, cette protéine favorise la formation d'un complexe contribuant aux processus de traduction au sein des ribosomes.
Symptômes Syndrome de Martin-Bell
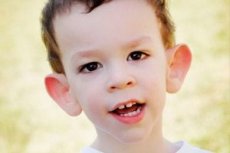
Comment reconnaître la maladie chez l'enfant? Quels sont les premiers signes? Dans les premiers mois de la vie d'un enfant, il est impossible de reconnaître le symptôme de Martin-Bell, si ce n'est qu'on observe parfois une diminution du tonus musculaire. Après un an, le tableau clinique est plus évident: l'enfant commence à marcher et à parler avec retard, parfois même à ne plus parler. Il est hyperactif, agite les bras au hasard, a peur de la foule et du bruit, est têtu, présente de violentes crises de colère, une instabilité émotionnelle, des crises d'épilepsie et ne regarde pas dans les yeux. Chez les patients atteints du syndrome de Martin-Bell, la maladie se révèle également par leur apparence: oreilles décollées et grandes, front lourd, visage allongé, menton proéminent, strabisme, mains et pieds larges. Ils sont également caractérisés par des troubles endocriniens: souvent un surpoids, une obésité, de gros testicules chez l'homme et une puberté précoce.

Parmi les patients atteints du syndrome de Martin-Bell, le niveau d'intelligence varie considérablement: d'un retard mental léger à des cas graves. Si une personne normale a un QI moyen de 100 et un génie de 130, les personnes susceptibles d'être atteintes de la maladie ont un QI compris entre 35 et 70.
Tous les symptômes cliniques de la pathologie peuvent être caractérisés par une triade de manifestations principales:
- oligophrénie (QI entre 35 et 50);
- dysmorphophobie (on observe des oreilles décollées et un prognathisme);
- macroorchidie, qui apparaît après le début de la puberté.
Environ 80 % des patients présentent également un prolapsus de la valve bicuspide.
Cependant, la forme complète du syndrome ne se manifeste que chez 60 % des patients. Chez 10 % d'entre eux, seul un retard mental est détecté, et chez les autres, la maladie se développe avec une combinaison différente de symptômes.
Parmi les premiers signes de la maladie, qui apparaissent dès le plus jeune âge:
- l'enfant malade présente un retard mental important par rapport au développement de ses pairs;
- troubles de l'attention et de la concentration;
- forte obstination;
- les enfants commencent à marcher et à parler assez tard;
- on observe des troubles de l’hyperactivité et du développement de la parole;
- accès de colère très forts et incontrôlables;
- un mutisme peut se développer – il s’agit d’une absence totale de parole chez un enfant;
- le bébé souffre d’anxiété sociale et est capable de paniquer à cause d’un bruit fort ou de tout autre son fort;
- l'enfant agite ses bras de manière incontrôlable et chaotique;
- on observe de la timidité, l'enfant a peur de se retrouver dans des endroits bondés;
- l'émergence de diverses idées obsessionnelles, état émotionnel instable;
- Le bébé peut être réticent à établir un contact visuel avec les gens.
Chez l'adulte, les symptômes suivants de la pathologie sont observés:
- aspect spécifique: un visage allongé avec un front lourd, de grandes oreilles décollées, un menton fortement saillant;
- pieds plats, otites et strabisme;
- la puberté survient assez tôt;
- l’obésité peut se développer;
- Assez souvent, des malformations cardiaques sont observées dans le syndrome de Martin-Bell;
- chez les hommes, on observe une hypertrophie des testicules;
- les articulations des articulations deviennent très mobiles;
- le poids et la taille augmentent fortement.
Diagnostics Syndrome de Martin-Bell
Pour diagnostiquer le syndrome de Martin Bell, il est nécessaire de consulter un généticien qualifié. Le diagnostic est posé après des tests génétiques spécifiques permettant d'identifier le chromosome défectueux.
Tests
À un stade précoce de la maladie, une méthode cytogénétique est utilisée: un fragment de matériel cellulaire est prélevé du patient, auquel on ajoute ensuite de l'acide folique pour provoquer des modifications chromosomiques. Après un certain temps, une zone du chromosome présentant un amincissement notable est identifiée, signe de la présence du syndrome de l'X fragile.
Cependant, ce test n’est pas adapté au diagnostic dans les stades avancés de la maladie, car sa précision est réduite par l’utilisation généralisée de multivitamines contenant de l’acide folique.
Le diagnostic intégré du syndrome de Martin-Bell est un examen génétique moléculaire, qui consiste à déterminer le nombre de répétitions dites trinucléotidiques dans le gène.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Diagnostic instrumental
Une méthode très spécifique de diagnostic instrumental est la PCR (réaction en chaîne par polymérase), qui permet d'étudier la structure des résidus d'acides aminés contenus dans le chromosome X et de déterminer ainsi la présence du syndrome de Martin Bell.
Il existe également une méthode distincte, encore plus spécifique, de diagnostic pathologique: une combinaison de PCR et de détection par électrophorèse capillaire. Cette méthode, très précise, permet de détecter les pathologies chromosomiques chez les patientes atteintes d'insuffisance ovarienne primaire et de syndrome ataxique.
La présence du défaut peut être déterminée après un diagnostic par EEG. Les patients atteints de cette maladie présentent une activité cérébrale bioélectrique similaire.
Quels tests sont nécessaires?
Diagnostic différentiel
Les méthodes différenciées qui aident à suspecter le syndrome comprennent:
- clinique - 97,5 % des patients présentent des signes évidents de retard mental (modéré ou profond); 62 % ont de grandes oreilles décollées; 68,4 % ont un menton et un front larges et proéminents; 68,4 % des garçons ont des testicules hypertrophiés, 41,4 % ont des particularités de la parole (débit de parole irrégulier, volume incontrôlable, etc.);
- cytogénique - le sang est examiné pour la culture des lymphocytes, le nombre de cellules avec un chromosome X fragile pour 100 cellules étudiées est déterminé;
- Électroencéphalographie - les modifications des impulsions électriques du cerveau spécifiques au syndrome de Martin-Bell sont enregistrées.
Qui contacter?
Traitement Syndrome de Martin-Bell
Chez les patients adultes, des antidépresseurs et des psychostimulants sont utilisés. Le traitement médicamenteux est suivi en permanence par un psychologue et un psychiatre. De plus, des micro-injections de médicaments tels que la cérébrolysine (ou ses dérivés) et les cytomédines (comme le Solcoseryl ou la Lidase) sont pratiquées en clinique privée.
Lors du développement du syndrome ataxique, des anticoagulants et des nootropes sont utilisés. Des mélanges d'acides aminés et des angioprotecteurs sont également prescrits. Les femmes présentant une insuffisance ovarienne primaire bénéficient d'un traitement correcteur à base de plantes médicinales et d'œstrogènes.
Les antagonistes des récepteurs de la glutamine sont également utilisés dans le traitement.
Traditionnellement, le traitement du syndrome de Martin-Bell repose sur l'utilisation de médicaments qui agissent sur les symptômes de la maladie, mais pas sur sa cause. Ce traitement comprend la prescription d'antidépresseurs, de neuroleptiques et de psychostimulants. Tous les médicaments ne sont pas indiqués chez l'enfant, la liste des médicaments disponibles est donc assez limitée. Les neuroleptiques utilisables après 3 ans (âge le plus précoce pour leur prescription) comprennent l'halopéridol en gouttes et en comprimés, la chlorpromazine en solution et la périciazine en gouttes. Ainsi, la dose d'halopéridol pour les enfants est calculée en fonction du poids corporel. Pour les adultes, la dose est prescrite individuellement. La dose est prise par voie orale, en commençant par 0,5 à 5 mg 2 à 3 fois par jour, puis la dose est progressivement augmentée jusqu'à 10 à 15 mg. En cas d'amélioration, la dose est réduite pour maintenir l'état initial. En cas d'agitation psychomotrice, 5 à 10 mg sont prescrits par voie intramusculaire ou intraveineuse, plusieurs fois après 30 à 40 minutes. La dose quotidienne ne doit pas dépasser 100 mg. Des effets secondaires tels que nausées, vomissements, spasmes musculaires, augmentation de la pression artérielle, arythmie, etc. sont possibles. Les personnes âgées doivent prendre des précautions particulières, car des cas d'arrêt cardiaque soudain ont été signalés et une dyskinésie tardive (mouvements involontaires) peut survenir.
Les antidépresseurs augmentent l'activité des structures cérébrales, soulagent la dépression, la tension et améliorent l'humeur. Ces médicaments, recommandés pour le syndrome de Martin-Bell entre 5 et 8 ans, comprennent la clomipromine, la sertraline, la fluoxétine et la fluvoxamine. La fluoxétine se prend par voie orale au cours des repas, une à deux fois (de préférence en début de journée), en commençant par 20 mg par jour, puis en augmentant à 80 mg si nécessaire. Il est déconseillé aux personnes âgées de dépasser 60 mg. La durée du traitement est déterminée par un médecin, mais ne doit pas dépasser 5 semaines.
Effets secondaires possibles: étourdissements, anxiété, acouphènes, perte d’appétit, tachycardie, œdème, etc. La prudence est de mise lors de la prescription aux personnes âgées, aux personnes atteintes de maladies cardiovasculaires et au diabète.
Les psychostimulants sont des médicaments psychotropes utilisés pour améliorer la perception des stimuli externes: ils aiguisent l'ouïe, les réactions de réponse et la vision.
Le diazépam est prescrit comme sédatif pour les névroses, l'anxiété, les crises d'épilepsie et les convulsions. Il est administré par voie orale, intraveineuse, intramusculaire ou rectale. Il est prescrit individuellement, selon la gravité de la maladie, à la dose minimale de 5 à 10 mg et à la dose quotidienne de 5 à 20 mg. La durée du traitement est de 2 à 3 mois. Chez l'enfant, la dose est calculée en fonction du poids corporel et des caractéristiques individuelles. Les effets secondaires incluent léthargie, apathie, somnolence, nausées et constipation. L'association avec l'alcool est dangereuse, une dépendance à ce médicament étant possible.
Dans le traitement du syndrome de Martin-Bell, des cas d'amélioration ont été observés grâce à l'introduction de médicaments à base de matière animale (cerveau): cérébrolysate, cérébrolysine et cérébrolysate-M. Les principaux composants de ces médicaments sont des peptides qui favorisent la production de protéines dans les neurones, compensant ainsi les déficits protéiques. La cérébrolysine est administrée sous forme de jet de 5 à 10 ml, le traitement comprenant 20 à 30 injections. Le médicament est prescrit aux enfants à partir d'un an, par voie intramusculaire, à raison de 1 à 2 ml par jour pendant un mois. Des administrations répétées sont possibles. Effets secondaires: fièvre; contre-indication chez la femme enceinte.
Des tentatives de traitement par l'acide folique ont été menées, mais seul le comportement s'est amélioré (diminution de l'agressivité et de l'hyperactivité, amélioration de la parole), et rien n'a changé au niveau intellectuel. Pour améliorer l'état de santé, l'acide folique est prescrit, des méthodes de physiothérapie, d'orthophonie, ainsi qu'une correction pédagogique et sociale sont indiquées.
Les préparations à base de lithium sont également considérées comme efficaces, car elles contribuent à améliorer l'adaptation du patient à l'environnement social et son activité cognitive. De plus, elles régulent son comportement en société.
L'utilisation de plantes médicinales pour le syndrome de Martin-Bell est possible comme antidépresseur. Parmi les plantes qui aident à soulager la tension, l'anxiété et à améliorer le sommeil, on trouve la valériane, la menthe poivrée, le thym, le millepertuis et la camomille. Les infusions se préparent comme suit: pour une cuillère à café de plantes sèches, il faut un verre d'eau bouillante; les décoctions sont infusées pendant au moins 20 minutes, principalement le soir avant le coucher ou l'après-midi. Une cuillerée de miel serait un bon complément.
Traitement de physiothérapie
Pour éliminer les manifestations neurologiques, des procédures physiothérapeutiques spéciales sont effectuées - telles que des exercices en piscine, une relaxation musculaire et de l'acupuncture.
Traitement chirurgical
La chirurgie plastique, qui vise à améliorer l'apparence du patient, constitue également une étape importante du traitement. Elle s'adresse aux membres, aux oreillettes et aux organes génitaux. La correction de la gynécomastie avec épispadias et d'autres défauts esthétiques est également pratiquée.
La prévention
La seule méthode de prévention de la maladie est le dépistage prénatal des femmes enceintes. Des examens spécifiques permettent une détection précoce de la pathologie, après quoi il est recommandé d'interrompre la grossesse. Une autre solution consiste à recourir à la FIV, qui peut permettre à l'enfant d'hériter d'un chromosome X sain.
La prévention dépend de la réapparition ou de l'hérédité de la mutation génétique. Pour cela, un diagnostic génétique moléculaire est réalisé. L'absence de « chromosome X fragile » chez les membres de la famille témoigne de la fraîcheur de la mutation, ce qui signifie que le risque d'avoir un enfant atteint du syndrome de Martin-Bell est très faible. Dans les familles où il y a des personnes malades, le test permettra d'éviter la récidive.
Prévoir
Le pronostic vital du syndrome de Martin-Bell est favorable, mais pas celui de la guérison. L'espérance de vie dépend de la gravité de la maladie et des anomalies associées. Le patient peut mener une vie normale. Dans les formes sévères du syndrome de Martin-Bell, les patients risquent une invalidité permanente.
Espérance de vie
Le syndrome de Martin Bell n'a pas d'impact négatif grave sur la santé, de sorte que l'espérance de vie de la plupart des personnes diagnostiquées avec cette pathologie ne diffère pas des indicateurs standard.