Expert médical de l'article

Nouvelles publications
Le syndrome de Martin-Bell
Dernière revue: 23.04.2024

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
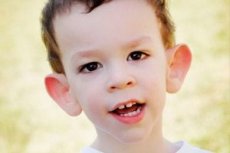
Le syndrome de Martin-Bella a été décrit en 1943. Médecins, dont les noms ont été nommés. La maladie est un trouble génétique, constitué d'un retard mental. En 1969 les changements caractéristiques de cette maladie sur le chromosome X (fragilité de l'épaule distale) ont été mis en évidence. En 1991 Les scientifiques ont découvert un gène responsable du développement de cette maladie. Cette maladie est également appelée le «syndrome du chromosome X fragile». La maladie affecte à la fois les garçons et les filles, mais le plus souvent (3 fois) les garçons sont malades.
 [
[Épidémiologie
Le syndrome de Martin-Bella est une maladie assez commune: il y a 0,3-1,0 personnes souffrant de cette maladie pour 1000 hommes, et 0,2-0,6 pour 1000 femmes. Et les enfants atteints du syndrome de Martin-Bell sont nés sur tous les continents avec la même fréquence. De toute évidence, la nationalité, la couleur de la peau, l'incision des yeux, les conditions de vie, le bien-être des personnes n'affectent pas l'apparition de la maladie. La fréquence de son apparition n'est comparable qu'avec la fréquence du syndrome de Down (pour 600 à 800 nouveau-nés, 1 maladie). Un cinquième des porteurs mâles du gène altéré sont en bonne santé, n'ont pas d'anomalies cliniques et génétiques, le reste présente des signes de retard mental de formes légères à sévères. Parmi les femmes porteuses de patients un peu plus d'un tiers.
Le syndrome du chromosome X fragile affecte environ 1 sur 2500-4000 hommes et 1 sur 7000-8000 femmes. La prévalence des porteurs de la maladie parmi la population féminine est estimée à 1 pour 130-250 personnes; la prévalence des porteurs chez les mâles est estimée à 1 pour 250-800.
Causes le syndrome de Martin-Bell
Le syndrome de Martin-Bella se développe en raison de l'arrêt complet ou partiel de la production d'une protéine spécifique par le corps. Cela est dû à l'absence de réponse d'un gène comme FMR1, localisé dans le chromosome X. La mutation se produit à la suite d'un réarrangement de la structure du gène à partir de variants structurels instables des états géniques (allèles), et non depuis le début. La maladie n'est transmise que par la ligne masculine, avec laquelle un homme n'est pas nécessairement malade. Les porteurs masculins, transmettent le gène à leurs filles sous forme inchangée, de sorte que leur retard mental n'est pas évident. Avec le transfert ultérieur du gène de la mère à ses enfants, le gène mute, puis tous les signes caractéristiques de cette maladie apparaissent.
Pathogénèse
Au coeur de la pathogenèse du syndrome de Martin-Bell se trouvent des mutations dans l'appareil génique qui conduisent au blocage de la production de la protéine FMR, une protéine vitale pour le corps, notamment dans les neurones, et présente dans divers tissus. Des études montrent que les protéines FMR sont directement impliquées dans la régulation des traductions qui se produisent dans les tissus cérébraux. L'absence de cette protéine ou sa production limitée par le corps conduit à un retard mental.
Dans la pathogenèse de la maladie, la violation clé est l'hyperméthylation du gène, mais il n'a pas été possible de déterminer finalement le mécanisme du développement de ce trouble.
Parallèlement à cela, l'hétérogénéité locus de la pathologie a également été révélée, ce qui est associé au polyallisme, ainsi que la polylocalisation. La présence de variants alléliques du développement de la maladie, qui sont causés par l'existence de mutations ponctuelles, ainsi que la destruction du gène FMRL, est déterminée.
Aussi chez les patients, 2 triplés fragiles sensibles à l'acide folique situés en Z00 kb sont détectés, ainsi que 1,5-2 mp. Du triplet fragile, qui contient le gène FMR1. Le mécanisme des gènes FRAXE, ainsi que les mutations FRAXF (ils sont identifiés dans les triplés triples précités) sont en corrélation avec le mécanisme des troubles du syndrome de Martin Bell. Ce mécanisme est dû à la propagation de GCC-, ainsi qu'aux répétitions CGG, sous lesquelles se produit la méthylation des îlots CpG. En plus de la forme classique de la pathologie, il existe également 2 types rares qui diffèrent en raison de l'expansion des répétitions trinucléotidiques (dans la méiose masculine et dans la méiose féminine).
Il a été révélé que dans la forme classique du syndrome, le patient ne possède pas de protéine nucléocytoplasmique spéciale du type FMR1, qui remplit la fonction de liaison d'une variété d'ARNm. En outre, cette protéine favorise la formation d'un complexe qui aide à effectuer des processus de traduction à l'intérieur du ribosome.
Symptômes le syndrome de Martin-Bell
Comment reconnaître la maladie chez les enfants? Quels sont les premiers signes? Dans les premiers mois de la vie de l'enfant, le symptôme de Martin-Bell ne peut être reconnu, sauf que parfois il y a une diminution du tonus musculaire. Après un an, la clinique de la maladie est plus évidente: l'enfant commence à marcher tard et à parler, parfois la parole est complètement absente. Il est hyperactif, agitant les bras au hasard, effrayé par la foule et le bruit, entêté, il y a de fortes explosions de colère, une instabilité émotionnelle, des crises d'épilepsie se produisent, ne vont pas au contact visuel. Chez les patients atteints du syndrome de Martin-Bella, la maladie produit également une apparence: les oreilles saillantes et grandes, le front lourd, le visage allongé, le menton saillant, le strabisme, les larges brosses et les pieds. Les troubles endocriniens sont également caractéristiques d'eux: poids souvent lourd, l'obésité, les hommes ont de grands testicules, la puberté précoce.

Parmi les patients atteints du syndrome de Martin-Bell, le niveau d'intelligence est très différent: d'un petit retard mental à ses cas sévères. Si le QI d'une personne normale est en moyenne de 100, et qu'un génie en a 130, alors les personnes touchées par la maladie en ont 35-70.
Tous les symptômes cliniques de la pathologie peuvent être caractérisés par une triade de manifestations fondamentales:
- l'oligophrénie (le QI est de 35 à 50);
- dysmorphophobie (oreilles protubérantes, ainsi que prognathisme);
- la macroorchidie, qui se manifeste après le début de la puberté.
Environ 80% des patients révèlent également le prolapsus de la valve bicuspide.
Mais la forme complète du syndrome ne se manifeste que chez 60% des patients. Dans 10% des cas seulement un retard mental est trouvé, tandis que dans d'autres la maladie se développe avec une combinaison différente de symptômes.
Parmi les premiers signes de la maladie, manifestés dès le plus jeune âge:
- l'enfant malade a un retard mental important par rapport au développement des autres pairs;
- troubles de l'attention et de la concentration;
- fort entêtement;
- les enfants commencent à marcher et à parler assez tard;
- il y a de l'hyperactivité et des perturbations dans le développement de la parole;
- des accès de colère très forts et incontrôlés;
- peut développer un mutisme - c'est l'absence complète du discours d'un enfant;
- l'enfant ressent de l'anxiété sociale, est capable de paniquer à cause d'un bruit fort ou de tout autre bruit fort;
- l'enfant de façon incontrôlable et chaotique agitant ses mains;
- il y a de la timidité, l'enfant a peur de rester dans les endroits d'une grande foule;
- l'émergence de diverses obsessions, un état émotionnel instable;
- le bébé peut être réticent à établir un contact visuel avec les gens.
Chez l'adulte, les symptômes de pathologie suivants sont observés:
- apparence spécifique: visage allongé avec front lourd, grandes oreilles saillantes, menton fortement saillant;
- pieds plats, otites et strabismes;
- la puberté se produit assez tôt;
- l'obésité peut se développer;
- assez souvent dans le syndrome de Martin Bell il y a des défauts dans le développement du coeur;
- chez les hommes, il y a une augmentation des testicules;
- les articulations des articulations deviennent très mobiles;
- Le poids, et aussi la croissance augmente fortement.
Diagnostics le syndrome de Martin-Bell
Pour diagnostiquer le syndrome de Martin Bell, vous devez contacter un généticien qualifié. Le diagnostic est fait après la réalisation de tests génétiques spécifiques, qui permettent de déterminer le chromosome défectueux.
Analyses
A un stade précoce du développement de la maladie, une méthode cytogénétique est utilisée, dans laquelle le patient prend un morceau de matériau cellulaire, auquel de l'acide folique est ensuite ajouté pour provoquer des changements dans les chromosomes. Au bout d'un certain temps, on découvre la région du chromosome, sur laquelle on observe un amincissement notable - c'est le signe de la présence du fragile syndrome du chromosome X.
Mais cette analyse ne convient pas pour le diagnostic dans les derniers stades de la maladie, car sa précision est réduite en raison de la large utilisation de multivitamines contenant de l'acide folique.
Le diagnostic intégré du syndrome de Martin-Bell est un examen génétique moléculaire, consistant à déterminer le nombre de répétitions dites trinucléotidiques dans le gène.
Diagnostic instrumental
Une méthode très spécifique de diagnostic instrumental est la PCR (Polymerase Chain Reaction), qui permet d'étudier la structure des résidus d'acides aminés contenus dans le chromosome X et de déterminer ainsi la présence du syndrome de Martin Bell.
Il existe également une méthode distincte, encore plus spécifique, pour diagnostiquer la pathologie - une combinaison de PCR et de détection à l'aide de l'électrophorèse capillaire. Cette méthode est très précise et révèle une pathologie chromosomique chez les patients présentant une forme primaire d'insuffisance ovarienne, ainsi que le syndrome ataxique.
Déterminer la présence d'un défaut peut être après le diagnostic sur l'EEG. Chez les patients atteints de cette maladie, une activité cérébrale bioélectrique similaire est observée.
Quels tests sont nécessaires?
Diagnostic différentiel
Les méthodes différenciées qui aident à suspecter un syndrome incluent:
- clinique - 97,5% des patients présentent des signes évidents de retard mental (modéré ou profond); 62% avaient de grandes oreilles protubérantes; 68,4% avaient un front proéminent et un front proéminent; dans 68,4% des garçons - les testicules sont élargis, dans 41,4% - les caractéristiques de la parole (le débit de la parole est inégal, le volume est incontrôlable, etc.);
- cytogénétique - le sang est examiné pour la culture des lymphocytes, le nombre de cellules avec la rupture du chromosome X pour 100 cellules étudiées est déterminé;
- électroencéphalographie - les changements dans les impulsions électriques du cerveau sont spécifiques au syndrome de Martin-Bell.
Qui contacter?
Traitement le syndrome de Martin-Bell
Les antidépresseurs avec psychostimulants sont utilisés dans le traitement des patients adultes. Le processus de pharmacothérapie est constamment surveillé par un psychologue et un psychiatre. En outre, les cliniques privées effectuent des procédures de micro-injection avec des médicaments tels que Cerebrolysin (ou ses dérivés), ainsi que des cytomédines (telles que Solcoseryl ou Lidase).
Avec le développement du syndrome ataxique, des médicaments qui diluent le sang, ainsi que des nootropiques, sont utilisés. De plus, des mélanges d'acides aminés et des angioprotecteurs sont prescrits. Les femmes présentant une forme primaire d'insuffisance ovarienne se voient prescrire un traitement correctif avec des phyto-médicaments et des œstrogènes.
En outre, les antagonistes des récepteurs de la glutamine sont utilisés dans le traitement.
Traditionnel pour le traitement du syndrome de Martin-Bell est l'utilisation de médicaments qui affectent les symptômes de la maladie, mais pas sur sa cause. Cette thérapie consiste en la prise d'antidépresseurs, de neuroleptiques, de psychostimulants. Tous les médicaments ne sont pas indiqués chez les enfants. La liste des médicaments est donc plutôt limitée. Les neuroleptiques qui peuvent être utilisés après 3 ans (l'âge le plus précoce de leur rendez-vous) comprennent l'halopéridol en gouttes et en comprimés, la chlorpromazine en solution, la péricyazine en gouttes. Ainsi, la dose de l'halopéridol pour les enfants est calculée en fonction du poids corporel. Pour les adultes, la dose est administrée individuellement. Acceptée à l'intérieur, commencez par 0,5-5 mg 2-3 fois par jour, puis augmentez graduellement la dose jusqu'à 10-15 mg. Quand il y a une amélioration, aller à une dose plus faible, pour maintenir l'état atteint. Avec la stimulation psychomotrice fixent 5-10 mg par voie intramusculaire ou intraveineuse, plusieurs répétitions en 30-40 minutes sont possibles. La dose quotidienne ne doit pas dépasser 100 mg. Les effets secondaires possibles sous la forme de nausées, vomissements, spasmes, augmentation de la pression, arythmie, etc. Des précautions spéciales doivent être suivies par les personnes âgées. Des cas d'arrêts cardiaques soudains ont été signalés et des discénérations tardives peuvent apparaître (apparition de mouvements involontaires).
Les antidépresseurs augmentent l'activité des structures cérébrales, soulagent l'humeur dépressive, la tension, augmentent l'humeur. Ces médicaments, recommandés pour l'admission de 5-8 ans avec le syndrome de Martin-Bell, incluent la clomipromine, la sertraline, la fluoxegine, la fluvoxamine. Ainsi, la fluoxétine est prise pendant les repas à l'intérieur de 1-2 (de préférence le matin), commencez avec 20 mg par jour, en augmentant jusqu'à 80 mg si nécessaire. Les personnes âgées ne recommandent pas une dose supérieure à 60 mg. Le cours du traitement est déterminé par le médecin, mais pas plus de 5 semaines.
Effets indésirables possibles: vertiges, anxiété, acouphènes, diminution de l'appétit, tachycardie, tuméfaction, etc. Des précautions doivent être prises lors de la nomination des personnes âgées, avec des maladies cardio-vasculaires, le diabète sucré.
Psychostimulants - médicaments psychotropes, utilisés pour améliorer la perception des stimuli externes: exacerber l'audition, la réponse, la vision.
Comme un sédatif avec des névroses, de l'anxiété, des crises d'épilepsie, des convulsions, le diazépam est prescrit. Il est pris par voie orale, intraveineuse, intramusculaire, rectale (dans le rectum). Il est prescrit individuellement, en fonction de la gravité de la maladie, à partir des plus petites doses de 5-10 mg, tous les jours - 5-20 mg. La durée du traitement est de 2-3 mois. Pour les enfants, la dose est calculée en tenant compte du poids corporel et des caractéristiques individuelles. Les effets secondaires incluent la léthargie, l'apathie, la somnolence, la nausée, la constipation. Il est dangereux de combiner avec de l'alcool, peut-être une dépendance à la drogue.
Dans le traitement du syndrome de Martin-Bell, des cas d'amélioration de la condition ont également été enregistrés lorsque des médicaments à base de matériel animal (cerveau) ont été fabriqués: cérébrolysat, cérébrolysine, cérébrolysat-M. Les principaux composants de ces médicaments sont des peptides, qui contribuent à la production de protéines dans les neurones, reconstituant ainsi la protéine manquante. Cerebrolysin injecté à 5-10 ml, le cours du traitement consiste en 20-30 injections. Les enfants prescrivent le médicament dès l'âge de la vie, injecté par voie intramusculaire tous les jours pour 1-2 ml au cours du mois. Séances répétées possibles d'admission. Les effets secondaires sous forme de chaleur, contre-indiqué chez les femmes enceintes.
Il y a eu des tentatives pour traiter la maladie avec de l'acide folique, mais seul l'aspect comportemental a été amélioré (le niveau d'agression, l'hyperactivité a diminué, la parole s'est améliorée), mais sur le plan intellectuel rien n'a changé. Pour améliorer l'état de la maladie prescrit l'acide folique, des méthodes de physiothérapie, correction logopédique, pédagogique et sociale est indiquée.
Les préparations au lithium sont également considérées comme efficaces, ce qui aide à améliorer l'adaptation du patient dans l'environnement social, ainsi que l'activité cognitive. En outre, ils régulent toujours son comportement dans la société.
L'utilisation d'herbes dans le syndrome de Martin-Bell est possible en tant qu'antidépresseurs. Les herbes qui aident à soulager la tension, l'anxiété, améliorer le sommeil comprennent la valériane, la menthe poivrée, le thym, le millepertuis, la camomille. Les infusions sont préparées comme suit: 1 cuillère à thé d'herbes séchées aura besoin d'un verre d'eau bouillante, les infusions insisteront au moins 20min, prises principalement la nuit avant d'aller se coucher ou dans l'après-midi. Un bon ajout à eux sera une cuillerée de miel.
Traitement physiothérapeutique
Pour éliminer les manifestations neurologiques, des procédures spéciales de physiothérapie sont pratiquées - telles que des exercices de piscine, la relaxation musculaire et l'acupuncture.
Traitement opératoire
Une étape importante du traitement est également considéré comme des méthodes de chirurgie plastique - opérations qui aident à améliorer l'apparence du patient. Plastique des extrémités et des oreillettes, et d'ailleurs ces organes génitaux sont effectués. Il y a aussi une correction de gynécomastie avec epispadia, et avec elle d'autres défauts d'apparence.
La prévention
La seule méthode de prévention de la maladie est le dépistage prénatal des femmes enceintes. Il existe des tests spéciaux qui permettent une détection précoce de la présence de la pathologie, après quoi il est recommandé d'interrompre la grossesse. Comme alternative, la FIV est utilisée pour aider un enfant à hériter d'un chromosome X sain.
La prophylaxie du patient dépend si la mutation du gène est ressortie ou a été héritée. Pour cela, le diagnostic génétique moléculaire est effectué. En faveur de la "fraîcheur" de la mutation, le fait que les proches n'aient pas révélé de "chromosome X fragile" témoigne du fait que le risque d'avoir un enfant atteint du syndrome de Martin-Bell est très faible. Dans les familles où il y a des patients, le test aidera à éviter les cas répétés.
Prévoir
Le pronostic du syndrome de Martin-Bell à vie est favorable, pour la guérison - non. L'espérance de vie dépend de la gravité de la maladie et des vices qui l'accompagnent. Le patient peut vivre une vie de durée habituelle. Dans les formes sévères du syndrome de Martin-Bell, une invalidité potentiellement mortelle menace les patients.
Durée de vie
Le syndrome de Martin Bell n'a pas d'impact négatif sérieux sur la santé, donc l'espérance de vie de la plupart des personnes qui ont trouvé cette pathologie ne diffère pas des indicateurs standards.