Expert médical de l'article

Nouvelles publications
L'idiotie amaurotique
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.

L'idiotie amaurotique est une maladie évolutive rare. Elle se caractérise par une baisse progressive de la vision jusqu'à la cécité complète et une dégradation de l'intelligence jusqu'à l'installation de l'idiotie. Le patient développe alors un marasme profond d'issue fatale. La maladie a été décrite pour la première fois par le Dr Tau, ophtalmologiste, il y a plus de 130 ans. Ce dernier a observé une transformation particulière du fond d'œil. Plus de 500 cas de cette maladie ont déjà été décrits.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
Épidémiologie
L'épidémiologie de la maladie révèle un caractère familial. Dans les familles où l'on retrouve des patients atteints d'idiotie amaurotique, les mariages consanguins sont dangereux pour la même raison. Les personnes d'origine canadienne-française ou juive sont également à risque.
[ 11 ], [ 12 ], [ 13 ], [ 14 ], [ 15 ], [ 16 ], [ 17 ], [ 18 ]
Causes idiotie amaurotique
Malgré les nombreuses données recueillies sur la maladie, les scientifiques continuent actuellement de chercher des réponses à de nombreuses questions sur les causes, la pathogenèse et même les manifestations de l'idiotie amaurotique.
Certains suggèrent que la maladie est héréditaire. Le mode de transmission est autosomique récessif. Le plus souvent, le cervelet et les lobes occipitaux des hémisphères cérébraux sont touchés, avec des conséquences graves et des complications pour l'ensemble de l'organisme: atrophie des nerfs optiques, perte possible des membranes des fibres nerveuses et désintégration des connexions entre les cellules nerveuses.
La plupart des experts reconnaissent que les signes cliniques de la maladie peuvent être très variés et sont en corrélation avec l’âge auquel l’idiotie amaurotique a commencé à se développer chez le patient.
Lors de l'étude des causes de la maladie, une certaine tendance a été observée: la maladie touche souvent les enfants d'une même famille, d'où le nom d'« idiotie amaurotique familiale ». Selon des études publiées au tout début de l'étude de la maladie, sur 64 cas d'idiotie amaurotique, 37 ont été recensés dans 13 familles (chaque famille comptait de 2 à 5 enfants malades). Il est à noter que dans ces familles, les malades avaient des frères et sœurs en parfaite santé. On pense aujourd'hui que le facteur de transmission récessive joue un rôle important dans le développement de la maladie. Ainsi, il est possible d'expliquer la fréquence de survenue de cas dans les mêmes familles. Lors de l'analyse du facteur héréditaire comme cause de l'idiotie amaurotique, il ne faut pas se limiter à la présence de signes cliniquement exprimés dans les familles de patients (tant dans les lignes ascendantes que latérales), mais également prendre en compte des signes rudimentaires, par exemple, des écarts caractéristiques dans le fonctionnement de l'appareil visuel (choroïdite familiale, dystrophie rétinienne pigmentaire, etc.).
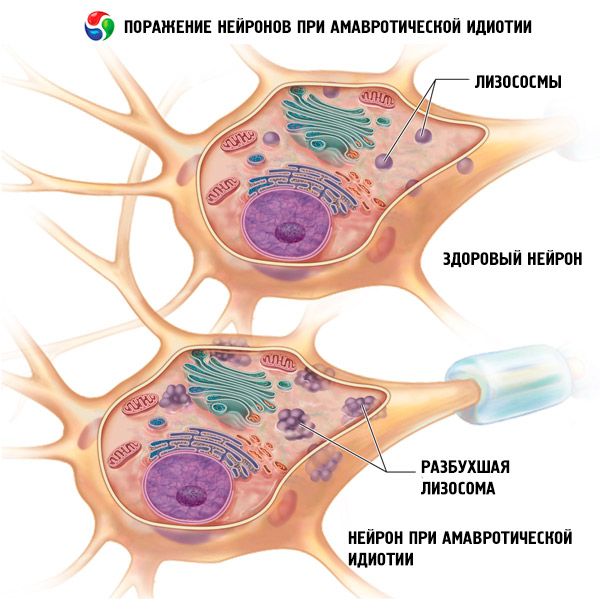
Symptômes idiotie amaurotique
Les premiers signes de la forme congénitale apparaissent dès les premiers jours ou semaines de vie. Le bébé naît avec une hydrocéphalie ou une microcéphalie, souffre de convulsions, de paralysie et de troubles respiratoires. Il décède après quelques mois.
Étapes
La forme infantile se développe entre 4 et 6 mois. Cette forme d'idiopathie amaurotique est caractérisée par un caractère familial. La vision décline rapidement: le bébé ne peut fixer son regard et n'observe pas les objets. Un « noyau de cerise » apparaît au fond de l'œil: une tache rougeâtre dans la région maculaire, entourée d'un bord gris-blanc. Les nerfs optiques s'atrophient ensuite et l'enfant perd complètement la vue. L'orientation, les réflexes de protection et la mobilité disparaissent progressivement. Les patients réagissent fortement aux stimuli sonores: ils sursautent à un son faible pour une personne en bonne santé; des convulsions peuvent survenir en raison d'une augmentation du tonus musculaire. Au stade terminal de la maladie, une atrophie générale, un épuisement du corps et une augmentation du tonus de tous les muscles extenseurs se développent. Le pronostic est également décevant: le patient décède un an et demi à deux ans après le début de la maladie.
La forme tardive de l'enfance débute vers 3-4 ans. La maladie progressive alterne avec des phases de rémission. La perte progressive de l'intelligence s'accompagne de convulsions, de troubles de la coordination et de troubles extrapyramidaux. Cette forme se caractérise également par une atrophie du nerf optique. Le décès survient 6 à 8 ans après le début de l'idiotie amaurotique.
La forme juvénile commence à se manifester entre 6 et 10 ans. L'idiotie amaurotique de Spielmeyer progresse moins rapidement. Les modifications du fond d'œil coïncident avec les manifestations d'une dystrophie rétinienne pigmentaire. La vision du patient décline progressivement, tout comme son intelligence. Les troubles moteurs peuvent se manifester de différentes manières et de manière irrégulière: une paralysie peu prononcée des bras et des jambes, ainsi que des troubles extrapyramidaux et bulbaires, peuvent survenir. La maladie entraîne le décès 10 à 25 ans après l'apparition des premiers signes.
La forme tardive est très rare et évolue extrêmement lentement. L'état mental du patient se modifie (comme dans un syndrome mental organique), une atrophie des nerfs optiques et une dystrophie pigmentaire de la rétine sont observées. Le stade final est caractérisé par une paralysie et un syndrome épileptiforme. Le patient décède 10 à 15 ans après le début de la maladie.
[ 38 ], [ 39 ], [ 40 ], [ 41 ], [ 42 ], [ 43 ], [ 44 ], [ 45 ]
Formes
Il existe quatre types d’idiotie amaurotique:
- Tay-Sachs (affectant dès le plus jeune âge);
- Jansky-Bilynovsky (apparaissant chez les enfants à un âge plus avancé);
- Syndrome de Spielmeyer-Vogt (survenant chez les adolescents);
- Kufsa (forme tardive).
Certains scientifiques distinguent également le type congénital Norman-Wood séparément.
Chaque type de maladie a son propre ensemble de manifestations cliniques, mais elles sont toutes unies par des causes communes, un tableau clinique, une base anatomique et une pathogenèse.
[ 46 ], [ 47 ], [ 48 ], [ 49 ], [ 50 ], [ 51 ], [ 52 ], [ 53 ], [ 54 ]
Diagnostics idiotie amaurotique
L'idiotie amaurotique est causée par un trouble du métabolisme lipidique, entraînant le dépôt d'un produit intermédiaire du métabolisme lipidique, la sphingomyéline, dans diverses cellules de l'organisme. La localisation et la composition de ces dépôts déterminent le développement d'un tableau clinique spécifique de la maladie.
[ 55 ], [ 56 ], [ 57 ], [ 58 ], [ 59 ], [ 60 ], [ 61 ], [ 62 ], [ 63 ]
Comment examiner?
Diagnostic différentiel
Le diagnostic différentiel de l'idiotie amaurotique repose sur un tableau clinique spécifique et des pathologies caractéristiques du fundus.
La forme précoce présente des symptômes similaires à ceux de la maladie de Landing, un type de mucopolysaccharidose. La maladie de Landing se développe dès les premiers mois suivant la naissance et entraîne le décès après 2 à 3 ans. Un « noyau de cerise » au fond d'œil apparaît dans 1/5 des cas; les modifications dégénératives de la rétine et les distorsions de la perception sonore (hypercussion) ne sont pratiquement pas caractéristiques de cette forme, mais on observe une hypertrophie simultanée de la rate et du foie, des troubles mentaux et des troubles du mouvement.
La forme juvénile se superpose parfois aux manifestations du syndrome de Lawrence-Moon-Biedl. Pour différencier ces maladies, il est nécessaire de prêter attention à leurs autres manifestations. Le syndrome de Lawrence-Moon-Biedl se caractérise par une prise de poids rapide, une déformation des membres caractérisée par la présence de doigts ou d'orteils supplémentaires, des troubles végétatifs-trophiques notables et l'absence de troubles de la fonction motrice.
La variété des symptômes de l'idiotie amaurotique tardive complique le diagnostic au cours de la vie. Ses manifestations sont similaires à celles de l'ataxie de Friedreich, de la sclérose en plaques, de la maladie d'Alzheimer, de la maladie de Pick, de la paralysie progressive et même de la schizophrénie.
Certains auteurs insistent sur le fait que le diagnostic de cette maladie, surtout lorsque les manifestations cliniques sont floues, ne peut être établi de manière fiable qu'après le décès, sur la base de l'analyse des anomalies histologiques du système nerveux.
Qui contacter?
Traitement idiotie amaurotique
Il n'existe aucun traitement rationnel et efficace. De nos jours, le traitement de l'idiotie amaurotique vise exclusivement à soulager les symptômes. On utilise des sédatifs, des nootropes, des anticonvulsivants et des toniques généraux.
Pour activer la circulation sanguine et les processus métaboliques dans le cerveau, la glycine, l'elkar, la cérébrolysine, l'acide glutamique et le pantogam sont prescrits.
Pour soulager le syndrome convulsif, on prescrit de la diphénine ou de la carmazépine.
Un résultat positif peut être obtenu en utilisant des extraits de tissus, une transfusion sanguine ou du plasma.
La prévention
L'absence de traitement efficace contre l'idiotie amaurotique nous oblige à accorder une attention particulière à la prévention. Il existe déjà des méthodes permettant d'identifier les porteurs hétérozygotes du gène pathologique et de diagnostiquer l'idiotie amaurotique pendant la grossesse. Le diagnostic prénatal de la maladie consiste à analyser l'activité de l'hexosaminidase A dans le liquide amniotique. Si une activité enzymatique réduite est détectée, il est recommandé d'interrompre la grossesse. Il est conseillé aux parents d'un enfant malade d'arrêter de procréer.
Использованная литература