Expert médical de l'article

Nouvelles publications
Encéphalomyopathie nécrosante subaiguë de Leah
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
La maladie a été mentionnée pour la première fois en 1951. À ce jour, plus de 120 cas ont été décrits. La maladie de Leigh (OMIM 256000) est une maladie génétiquement hétérogène qui peut être transmise soit par voie nucléaire (autosomique récessive ou liée à l'X), soit par voie mitochondriale (moins fréquente).
 [
[ Causes du syndrome de Leah
La maladie repose sur un déficit en enzymes assurant la production d'énergie, principalement dû à une perturbation du métabolisme de l'acide pyruvique et à un défaut de transport d'électrons dans la chaîne respiratoire. Un déficit du complexe pyruvate déshydrogénase (sous-unité α-E1), de la pyruvate carboxylase, du complexe 1 (NAD-coenzyme Q-réductase) et du complexe 4 (cytochrome oxydase) de la chaîne respiratoire se développe.
Il a été établi que les anomalies de la pyruvate carboxylase, du complexe 1 (NAD-coenzyme Q-réductase) et du complexe 4 (cytochrome oxydase) de la chaîne respiratoire sont transmises selon un mode autosomique récessif, tandis que celles du complexe pyruvate déshydrogénase (sous-unité a-E1) sont transmises selon un mode récessif lié à l'X. En cas de mutations ponctuelles de l'ADNmt, affectant la 6e sous-unité de l'ATPase, l'hérédité mitochondriale est typique. Le plus souvent, une mutation miscens survient, associée au remplacement de la thymine par la guanine ou la cytosine en position 8993 de l'ADNmt. Plus rarement, une mutation en position 9176 de l'ADNmt est observée. La mutation T8993G étant le principal défaut du syndrome NARP, des familles atteintes de ces deux maladies ont été décrites. Chez les enfants, une mutation de l'ADNmt en position 8344 a également été décrite, qui survient dans le syndrome MERRF.
On suppose qu'en cas d'accumulation d'ADNmt mutant dans la plupart des mitochondries, une forme sévère du syndrome de Leigh se développe. Dans la genèse mitochondriale de cette maladie, l'ADNmt mutant est présent dans 90 % des mitochondries. La pathogenèse est associée à une perturbation de la production d'énergie dans les cellules et au développement d'une acidose lactique.
Symptômes du syndrome de Leah
Les premiers signes de la maladie apparaissent dès le plus jeune âge (1 à 3 ans). Cependant, des cas de manifestation de la maladie ont été signalés à 2 semaines et à 6-7 ans. Au début, des troubles non spécifiques se développent: retard psychomoteur, diminution de l’appétit, épisodes de vomissements, déficit pondéral. Par la suite, les symptômes neurologiques s’aggravent: hypotonie musculaire ou dystonie avec transition vers l’hypertonie, crises de myoclonies ou crises tonico-cloniques, tremblements des extrémités, choréoathétose, troubles de la coordination, diminution des réflexes tendineux, léthargie, somnolence. La neurodégénérescence cérébrale est progressive. Les symptômes d’insuffisance pyramidale et extrapyramidale s’aggravent, la déglutition est altérée. Des modifications de la vision telles que ptosis, ophtalmoplégie, atrophie des nerfs optiques et, plus rarement, dégénérescence pigmentaire de la rétine sont souvent observées. Une cardiomyopathie hypertrophique se développe parfois, avec des épisodes de tachypnée.
Dans de rares cas, la maladie évolue vers une encéphalopathie aiguë. L'évolution chronique ou subaiguë est plus fréquente, entraînant le décès plusieurs années après le début de la maladie. En cas d'évolution rapide (plusieurs semaines), la mort survient par paralysie du centre respiratoire.
Diagnostics du syndrome de Leah
Un test sanguin biochimique révèle une acidose lactique due à l'accumulation d'acides lactique et pyruvique dans le sang et le liquide céphalorachidien, ainsi qu'à une augmentation du taux d'alanine dans le sang. Le taux de corps cétoniques peut également être élevé. Une augmentation de l'excrétion d'acides organiques est détectée dans les urines: acide lactique, acide fumarique, etc. Le taux de carnitine dans le sang et les tissus diminue souvent.
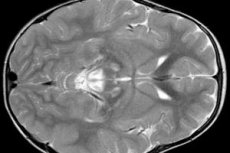
L'EEG révèle des signes focaux d'activité épileptique. L'IRM révèle une hypertrophie des ventricules cérébraux, des lésions cérébrales bilatérales et une calcification des noyaux gris centraux (noyau caudé, putamen, substance noire, globus pallidus). Une atrophie des hémisphères cérébraux et de la matière cérébrale peut également être détectée.
L'examen morphologique révèle des modifications importantes de la substance cérébrale: foyers symétriques de nécrose, démyélinisation et dégénérescence spongieuse du cerveau, principalement des sections moyennes, du pont, des noyaux gris centraux, du thalamus et du nerf optique. Le tableau histologique comprend une dégénérescence kystique du tissu cérébral, une gliose astrocytaire, une mort neuronale et une augmentation du nombre de mitochondries dans les cellules. Dans les muscles squelettiques, on observe une accumulation d'inclusions lipidiques, une diminution de la réaction histochimique aux complexes 1 et 4 de la chaîne respiratoire, une accumulation sous-sarcolemmale de mitochondries, des mitochondries anormales avec désorganisation des crêtes. Le phénomène de RRF est souvent passé inaperçu.
Comment examiner?
Quels tests sont nécessaires?
Использованная литература