Expert médical de l'article

Nouvelles publications
Encéphalomyopathie nécrosante subaiguë Leia
Dernière revue: 23.04.2024

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
 [
[Causes du syndrome de Leia
La maladie est basée sur une déficience enzymatique, fournissant de l'énergie de formation principalement par des troubles métaboliques de l'acide pyruvique et de transport d'électrons de défauts dans la chaîne respiratoire. Déficit en complexe pyruvate déshydrogénase se développe (a-E1 sous-unité), la pyruvate carboxylase, complexe 1 (NAD coenzyme Q-réductase) et complexe 4 (cytochrome oxydase), la chaîne respiratoire.
Il a été constaté que les défauts de pyruvate, complexe 1 (NAD coenzyme Q-réductase) et complexe 4 (cytochrome oxydase), la chaîne respiratoire sont hérités d'une manière autosomique récessive, des anomalies du complexe pyruvate déshydrogénase (a-E1 sous-unité) - récessive liée à l'X. Lorsque des mutations ponctuelles qui affectent la sous-unité ADNmt 6-ATPase, caractéristique de l'héritage mitochondrial. La plupart se produit souvent mutation de mistsens associés au remplacement de la thymine en guanine ou cytosine en position 8993 ADNmt. Moins commun est une mutation à la position 9176 ADNmt. En raison du fait que les mutations T8993G - défaut de base dans le syndrome NARP, décrit dans la famille avec la présence de ces deux maladies. Les enfants ont également été décrits mutation ADNmt à la position 8344, qui se trouve dans le syndrome merrf.
Il est suggéré que dans le cas de l'accumulation d'ADNmt mutant dans la majorité des mitochondries se développe une évolution sévère du syndrome de Leia. Dans la genèse mitochondriale de cet état, l'ADN mitochondrial mutant est détecté dans 90% de toutes les mitochondries. La pathogenèse est associée à une violation de la production d'énergie dans les cellules et au développement de l'acidose lactique.
Symptômes du syndrome de Leia
Les premiers signes de la maladie débuts à un âge précoce (1-3 ans). Cependant, les cas de manifestation de la maladie chez les enfants de 2 semaines et 6-7 ans sont connus. Dans un premier temps développé des troubles non spécifiques: un retard psychomoteur, perte d'appétit, vomissements, insuffisance pondérale épisodes. Dans les symptômes neurologiques suivants: croissance hypotonie musculaire ou dystonie avec la transition vers hypertonicité, des convulsions, myoclonies ou des crises tonico-cloniques, tremblements des membres, choréoathétose, troubles de la coordination, réflexes tendineux diminué, la léthargie, la somnolence. La neurodégénérescence cérébrale a une nature progressive. Les symptômes d'insuffisance pyramidale et extrapyramidale se développent, l'acte de déglutition est rompu. Souvent, il y a un tel changement d'autorité comme ptosis, ophtalmoplégie, atrophie optique, rétinite pigmentaire moins. Parfois, une cardiomyopathie hypertrophique se développe, des épisodes de tachypnée apparaissent.
Rarement, la maladie se déroule selon le type d'encéphalopathie aiguë. Plus caractéristique est un courant chronique ou subaiguë, qui conduit à une issue fatale quelques années après le début de la maladie. Avec un flux rapide (plusieurs semaines), la mort survient à la suite d'une paralysie du centre respiratoire.
Diagnostics du syndrome de Leia
Dans un test sanguin biochimique, l'acidose lactate est détectée en raison de l'accumulation d'acides lactiques et pyruviques dans le sang et la liqueur, ainsi que d'une augmentation de la teneur en alanine dans le sang. En outre, le niveau de corps cétoniques peut être augmenté. Dans l'urine, il y a une augmentation de l'excrétion des acides organiques: lactique, fumarique, etc. Le taux de carnitine dans le sang et les tissus est souvent réduit.
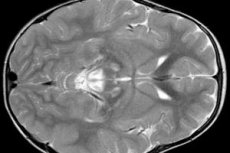
Les résultats de l'EEG révèlent des signes focaux de l'activité épileptique. Selon les données de l'IRM, une expansion des ventricules du cerveau, des lésions cérébrales bilatérales, une calcification des ganglions de la base (noyau caudé, coquillage, substance noire, boule pâle) sont détectées. Il est également possible d'identifier l'atrophie des hémisphères cérébraux et des substances cérébrales.
étude montrent des changements morphologiques bruts en substance cérébrale: nécrose symétrique, une démyélinisation et une dégénérescence spongieuse du cerveau, principalement des pièces intermédiaires, pont, noyaux gris centraux, le thalamus, le nerf optique. L'image histologique comprend la dégénérescence kystique du tissu cérébral, la gliose astrocytaire, la mort des neurones, une augmentation du nombre de mitochondries dans les cellules. Dans le muscle squelettique - l'accumulation d'inclusions lipidiques diminuer la réaction histochimique à des complexes de 1, 4 chaîne respiratoire de la congestion mitochondries de subsarkolemmalnoe, les mitochondries anormales avec la perturbation de cristae. Le phénomène de RRF n'est souvent pas détecté.
Comment examiner?
Quels tests sont nécessaires?
Использованная литература