Expert médical de l'article

Nouvelles publications
Lymphomes à cellules T de la peau
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
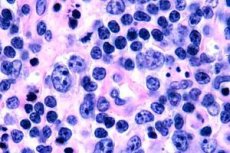
Le plus souvent, les lymphomes à cellules T sont observés chez les personnes âgées, bien que des cas isolés soient observés même chez les enfants. Les hommes sont deux fois plus touchés que les femmes. Les lymphomes à cellules T sont de nature épidermotrope.
Causes Lymphomes à cellules T de la peau
Les causes et la pathogenèse des lymphomes cutanés à cellules T ne sont pas entièrement comprises. Actuellement, la plupart des chercheurs considèrent le virus de la leucémie humaine à cellules T de type 1 (HTLV-1) comme le principal facteur étiologique à l'origine du développement des lymphomes cutanés malins à cellules T. Le rôle d'autres virus dans le développement des lymphomes à cellules T est également évoqué: le virus d'Epstein-Barr et l'herpès simplex de type 6. Chez les patients atteints de lymphome à cellules T, les virus sont présents dans la peau, le sang périphérique et les cellules de Langerhans. Des anticorps anti-HTLV-1 sont détectés chez de nombreux patients atteints de mycosis fongoïde.
Une place importante dans la pathogenèse des lymphomes à cellules T est jouée par les processus immunopathologiques de la peau, dont le principal est la prolifération incontrôlée de lymphocytes clonaux.
Les cytokines produites par les lymphocytes, les cellules épithéliales et les cellules du système macrophagique ont des effets pro-inflammatoires et prolifératifs (IL-1, responsable de la différenciation lymphocytaire; IL-2, facteur de croissance des lymphocytes T; IL-4 et IL-5, augmentant l'afflux d'éosinophiles dans la lésion et leur activation, etc.). Suite à l'afflux de lymphocytes T dans la lésion, des micro-abcès de Pautrier se forment. Simultanément à l'augmentation de la prolifération lymphocytaire, l'activité des cellules de défense antitumorales est supprimée: cellules tueuses naturelles, lymphocytes lymphocytotoxiques, cellules dendritiques, en particulier les cellules de Langerhans, ainsi que les cytokines (IL-7, IL-15, etc.) – inhibiteurs de la croissance tumorale. Le rôle de facteurs héréditaires ne peut être exclu. La présence de cas familiaux, la détection fréquente de certains antigènes d'histocompatibilité (HLA B-5 et HLA B-35 - dans les lymphomes cutanés hautement malins, HLA A-10 - dans les lymphomes moins agressifs, HLA B-8 - dans la forme érythrodermique du mycosis fongoïde) confirment le caractère héréditaire de la dermatose.
Les observations cliniques indiquent une possible transformation des dermatoses chroniques à long terme (neurodermatite, dermatite atopique, psoriasis, etc.) en mycosis fongoïde. Le facteur clé est la persistance prolongée des lymphocytes dans le foyer inflammatoire, ce qui perturbe la surveillance immunitaire et favorise l'émergence d'un clone de lymphocytes malins et, par conséquent, le développement d'un processus prolifératif malin.
L’impact de facteurs physiques sur le corps, tels que l’insolation, les rayonnements ionisants et les substances chimiques, peut conduire à l’émergence d’un clone de lymphocytes « génotraumatiques » qui ont un effet mutagène sur les cellules lymphoïdes et au développement de tumeurs malignes des lymphocytes.
Par conséquent, les lymphomes à cellules T peuvent être considérés comme une maladie multifactorielle qui débute par l'activation des lymphocytes sous l'influence de divers facteurs cancérigènes et « génotraumatisants » et l'émergence d'un clone dominant de cellules T. La gravité du trouble de la surveillance immunitaire, le clone de lymphocytes malins déterminent les manifestations cliniques (taches, plaques ou éléments tumoraux) des lymphomes à cellules T.
Pathogénèse
Au stade précoce du mycosis fongoïde, on observe une acanthose avec de larges prolongements, une hyperplasie et une compaction des kératinocytes basaux, une dégénérescence vacuolaire de certaines cellules basales, des mitoses atypiques dans différentes couches de l'épiderme, un épidermotropisme de l'infiltrat avec pénétration des lymphocytes dans l'épiderme. Dans le derme, on observe de petits infiltrats autour des vaisseaux, constitués de cellules mononucléaires uniques à noyaux hyperchromiques – cellules « mycotiques ». Au deuxième stade, on observe une augmentation de la sévérité de l'infiltrat dermique et un épidermotropisme des cellules infiltrées, entraînant la pénétration des lymphocytes malins dans l'épiderme, formant des amas sous forme de micro-abcès de Potrier. Au troisième stade, tumoral, on observe une acanthose massive et une légère atrophie de l'épiderme, ainsi qu'une infiltration accrue de l'épiderme par les lymphocytes tumoraux, qui forment de multiples micro-abcès de Potrier. L'infiltrat massif est localisé sur toute l'épaisseur du derme et recouvre une partie de l'hypoderme. On observe des lymphocytes de forme blastique.
Lymphome cutané anaplasique à grandes cellules T
Il s'agit d'un groupe de processus lymphoprolifératifs caractérisés par la présence de proliférations de lymphocytes T anaplasiques CD30+ clonaux atypiques de grande taille. En règle générale, il se développe secondairement au stade tumoral du mycosis fongoïde ou du syndrome de Sézary, mais peut se développer indépendamment ou avec la dissémination de lymphomes systémiques de ce type. Cliniquement, ces lymphomes correspondent à la forme dite décapitée du mycosis fongoïde, se présentant sous forme de ganglions uniques ou multiples, généralement groupés.
Histologiquement, le proliféré occupe la quasi-totalité du derme avec ou sans épidermotropisme en cas d'atrophie épidermique.
Cytologiquement, les cellules tumorales peuvent varier en taille et en forme. Sur la base de ces propriétés, on distingue les lymphomes T pléomorphes à cellules moyennes et à grandes cellules, dont les noyaux présentent diverses configurations irrégulières: convolutés, multilobés, avec une chromatine dense, un nucléole bien défini et un cytoplasme assez abondant; les lymphomes immunoblastiques, dont les noyaux sont de grande taille, ronds ou ovales, avec un caryoplasme clair et un nucléole central; les lymphomes anaplasiques, dont les cellules sont de très grande taille, disgracieuses, avec des noyaux de configuration irrégulière et un cytoplasme abondant. Phénotypiquement, ce groupe appartient aux lymphomes T auxiliaires et peut être CD30+ ou CD30-.
R. Willemze et al. (1994) ont montré que l'évolution du lymphome CD30+ est plus favorable. Génotypiquement, un réarrangement clonal du récepteur des lymphocytes T est détecté.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Symptômes Lymphomes à cellules T de la peau
La maladie la plus fréquente dans le groupe des lymphomes cutanés à cellules T est le mycosis fongoïde, qui représente environ 70 % des cas. Il existe trois formes cliniques: classique, érythrodermique et décapitée. Les lymphomes à cellules T se caractérisent par un polymorphisme d'éruptions cutanées sous forme de taches, de plaques et de tumeurs.
La forme érythrodermique du mycosis fongoïde débute généralement par des démangeaisons incontrôlables, un gonflement, une hyperémie généralisée et l'apparition de lésions érythémateuses-squameuses sur la peau du tronc et des extrémités, qui tendent à fusionner et à évoluer vers une érythrodermie en un à deux mois. Presque tous les patients présentent une hyperkératose palmo-plantaire et une perte diffuse de cheveux sur l'ensemble de la peau. Tous les groupes de ganglions lymphatiques sont fortement hypertrophiés. Les ganglions inguinaux, fémoraux, axillaires et cubitaux hypertrophiés sont palpés comme des « paquets » de consistance élastique dense, non fusionnés aux tissus environnants et indolores. L'état général se dégrade brutalement: fièvre avec une température corporelle pouvant atteindre 38-39 °C, sueurs nocturnes, faiblesse et perte de poids apparaissent. Actuellement, le syndrome de Sézary est considéré par de nombreux dermatologues comme la variante leucémique la plus rare de la forme érythrodermique du mycosis fongoïde.
Une leucocytose prononcée est observée sur les lymphocytogrammes: cellules de Sézary. Les cellules de Sézary sont des lymphocytes T auxiliaires malins, dont les noyaux présentent une surface cérébriforme repliée avec des invaginations profondes de la membrane nucléaire. L'issue fatale est observée après 2 à 5 ans, la cause fréquente étant une pathologie cardiovasculaire et une intoxication.
La forme décapitée du mycosis fongoïde se caractérise par le développement rapide de lésions tumorales sur une peau apparemment saine, sans formation préalable de plaques à long terme. Cette forme se caractérise par un degré élevé de malignité, considéré comme une manifestation de lymphosarcome. L'issue fatale est observée en moins d'un an.
Étapes
La forme classique du mycosis fongoïde est caractérisée par trois stades de développement: érythémateux-squameux, en plaques et tumoral.
Le premier stade ressemble au tableau clinique de certaines dermatoses inflammatoires bénignes: eczéma, dermatite séborrhéique, parapsoriasis en plaques. À ce stade de la maladie, on observe des plaques de différentes tailles, rose intense, rose-rouge avec une teinte violette, aux contours ronds ou ovales, aux limites relativement nettes, et présentant une desquamation superficielle en forme de son ou de fines plaques. Ces plaques sont souvent localisées sur différentes zones de la peau, le plus souvent sur le tronc et le visage. Leur nombre augmente progressivement. Avec le temps, le processus peut évoluer vers une érythrodermie (stade érythrodermique). L'éruption peut persister pendant des années ou disparaître spontanément. Contrairement aux dermatoses inflammatoires bénignes, les éléments de l'éruption et les démangeaisons à ce stade résistent au traitement.
Le stade des plaques infiltrantes se développe sur plusieurs années. À la place des éruptions cutanées tachetées préexistantes, apparaissent des plaques aux contours ronds ou irréguliers, d'un violet intense, clairement délimitées de la peau saine, denses et à la surface squameuse. Leur consistance ressemble à du carton épais. Certaines disparaissent spontanément, laissant des zones d'hyperpigmentation brun foncé et/ou d'atrophie (poïkilodermie). À ce stade, les démangeaisons sont encore plus intenses et douloureuses, de la fièvre et une perte de poids sont observées. Une lymphadénopathie peut également être observée à ce stade.
Au troisième stade tumoral, apparaissent des tumeurs indolores, denses et élastiques, de couleur jaune-rouge, se développant à partir de plaques ou apparaissant sur une peau apparemment saine. De forme sphérique ou aplatie, elles ressemblent souvent à un chapeau de champignon. Elles peuvent apparaître n'importe où. Leur nombre varie considérablement, d'une seule à plusieurs dizaines, et leur taille varie de 1 à 20 cm de diamètre. La désintégration des tumeurs anciennes entraîne la formation d'ulcères aux bords irréguliers et au fond profond, atteignant le fascia ou l'os. Les ganglions lymphatiques, la rate, le foie et les poumons sont le plus souvent touchés. L'état général s'aggrave, les symptômes d'intoxication apparaissent et s'intensifient, et une faiblesse se développe. L'espérance de vie moyenne des patients atteints de la forme classique de mycosis fongoïde, à compter du diagnostic, est de 5 à 10 ans. La mortalité est généralement observée en raison de maladies intercurrentes: pneumonie, insuffisance cardiovasculaire, amylose. Des démangeaisons sont ressenties subjectivement et, lors de la désintégration des tumeurs, des douleurs dans les zones touchées apparaissent.
Qu'est-ce qu'il faut examiner?
Comment examiner?
Traitement Lymphomes à cellules T de la peau
Au stade érythémateux-squameux, les patients n'ont pas besoin de traitement antitumoral; on leur prescrit des corticoïdes topiques (prednisolone, bétaméthasone, dérivés de la dexaméthasone), de l'interféron alpha (3 millions d'UI par jour, puis 3 fois par semaine pendant 3 à 6 mois selon les manifestations cliniques ou l'efficacité du traitement), de l'interféron gamma (100 000 UI par jour pendant 10 jours, le cycle étant répété 12 à 3 fois avec une pause de 10 jours), une PUVAthérapie ou une Re-PUVAthérapie. L'efficacité de la PUVAthérapie repose sur la formation sélective de liaisons covalentes des psoralènes avec l'ADN des lymphocytes T auxiliaires en prolifération, ce qui inhibe leur division. Au deuxième stade, en plus des agents mentionnés ci-dessus, on utilise des corticoïdes systémiques (30 à 40 mg de prednisolone par jour pendant 1,5 à 2 mois) et des cytostatiques (prospédine 100 mg par jour, 4 à 5 injections au total). L'association d'interférons à d'autres traitements (interférons + PUVA, interférons + cytostatiques, interférons + rétinoïdes aromatiques) a un effet thérapeutique plus marqué.
Au stade tumoral, la principale méthode est la polychimiothérapie. On utilise une association de vincristine (0,5 à 1 mg par voie intraveineuse une fois par jour, soit 4 à 5 injections au total), de prednisolone (40 à 60 mg par jour par voie orale pendant la chimiothérapie), de prospidine (100 mg par jour, soit 3 g au total) et d'interférons. La photothérapie dynamique, la thérapie par faisceau d'électrons et la photophérèse (photochimiothérapie extracorporelle) sont recommandées.