Expert médical de l'article

Nouvelles publications
L'alcaptonurie est une anomalie enzymatique congénitale.
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
L'alcaptonurie, l'un des troubles métaboliques les plus rares, fait référence à des anomalies congénitales du métabolisme de l'acide aminé tyrosine.
Ce syndrome peut également être appelé déficit en homogentisate oxydase, homogentisinurie, ochronose héréditaire ou maladie des urines noires.[ 1 ]
Épidémiologie
Selon les statistiques, on ne recense pas plus de neuf cas d'alcaptonurie pour un million de personnes. Dans la plupart des pays européens, on compte un cas pour 100 000 à 250 000 naissances vivantes.
Parmi les pays européens, la Slovaquie fait exception (notamment dans sa région relativement petite du nord-ouest), où la prévalence de l'alcaptonurie est d'un cas pour 19 000 nouveau-nés. Cela s'explique probablement par le fait que parmi les familles roms slovaques qui y vivent, le taux de consanguinité (mariages entre cousins) est le plus élevé d'Europe: 10 à 14 %. [ 2 ]
Causes alcaptonurie
Les causes exactes de l'alcaptonurie, trouble congénital du catabolisme (dégradation métabolique) de la tyrosine, un acide α-aminé aromatique (homocyclique), ont été établies: ce type de trouble métabolique est la conséquence de mutations homozygotes ou hétérozygotes composites de l'un des milliers de gènes du chromosome 3, plus précisément du gène HGD, situé au locus 3q21-q23 sur le bras long du chromosome. Ce gène code les séquences nucléotidiques de l'enzyme hépatique homogentisate-1,2-dioxygénase [ 3 ] (également appelée acide homogentisique oxydase ou homogentisate oxydase), une métalloprotéine contenant du fer, nécessaire à l'une des étapes de la dégradation de la tyrosine dans l'organisme. [ 4 ], [ 5 ]
Ainsi, l'alcaptonurie est un défaut de l'enzyme homogentisate-1,2-dioxygénase, ou plus précisément, le résultat de son déficit ou de son absence complète génétiquement déterminé. [ 6 ]
Étant une déficience enzymatique congénitale, l'alcaptonurie est héritée selon un trait autosomique récessif, c'est-à-dire que pour que l'alcaptonurie se produise chez les enfants, les deux parents doivent avoir un gène modifié pour l'enzyme, puisque chacun d'eux ne transmet à l'enfant qu'une seule copie du gène parmi les deux disponibles.
Selon les dernières données, il existe plus de deux cents variantes de modification du gène HGD, et les mutations faux-sens, la translocation et l'épissage sont les plus souvent observés.
Facteurs de risque
Le seul facteur de risque de développer cette enzymopathie congénitale est sa présence dans les antécédents familiaux et l'hérédité de deux copies modifiées du gène HGD, si les parents ne présentent pas d'alcaptonurie (le risque de transmettre l'anomalie est de 25%), ou si l'un des parents est atteint de ce trouble. [ 7 ]
Pathogénèse
La tyrosine joue un rôle clé dans la synthèse des protéines, la production de chromoprotéines – le pigment de la peau, la mélanine, ainsi que des hormones thyroïdiennes et des neurotransmetteurs catécholamines.
Le mécanisme de régulation de la quantité de tyrosine dans les cellules est très complexe, et l'organisme normalise son excès en la décomposant. Le catabolisme de la tyrosine, comme celui de tous les acides aminés aromatiques, se déroule en plusieurs étapes. Chaque étape de la dégradation métabolique de la tyrosine implique une enzyme spécifique et la formation d'un composé intermédiaire.
Ainsi, l'acide aminé est d'abord décomposé en para-hydroxyphénylpyruvate, qui est ensuite converti en acide alcaptone – acide 2,5-dihydroxyphénylacétique ou acide homogentisique. L'alcaptone devrait ensuite être transformée en acide maléacétique, mais cela n'a pas lieu. [ 8 ]
Et la pathogénèse de l'alcaptonurie consiste en l'arrêt des réactions biochimiques du catabolisme de la tyrosine au stade de la formation de l'acide homogentisique: il n'y a tout simplement pas d'enzyme nécessaire pour le décomposer – l'homogentisate oxydase.
L'acide homogentisique n'est pas utilisé par l'organisme et peut s'accumuler lors de son excrétion rénale. De plus, il est oxydé en benzoquinoacétate (acide benzoquinoneacétique), qui, en se liant aux molécules des tissus et des fluides corporels, forme des composés biopolymères colorés comme la mélanine.
L'accumulation de ces produits intermédiaires dans le tissu entraîne une perturbation de la structure collagène du tissu cartilagineux, ce qui réduit son élasticité – avec l'apparition de nombreux signes cliniques d'alcaptonurie et le développement de complications.
Symptômes alcaptonurie
L'alcaptonurie chez les nouveau-nés et les nourrissons se caractérise par une coloration foncée de l'urine. Exposée à l'air, l'urine présente sur les couches, les langes et les sous-vêtements prend une teinte brun foncé; ce phénomène est dû à l'accumulation et à la libération d'acide homogentisique, oxydé en benzoquinoacétate. [ 9 ]
En l'absence d'autres symptômes, l'alcaptonurie chez les jeunes enfants n'est souvent pas diagnostiquée à temps, car l'urine peut devenir foncée après plusieurs heures de miction. Selon certaines données, seul un cinquième des enfants de moins de 12 mois nés avec ce déficit enzymatique sont diagnostiqués en milieu clinique. Il est donc essentiel que les parents soient attentifs aux soins prodigués à leurs nourrissons.
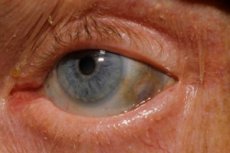
De plus, les premiers signes comprennent une pigmentation (couleur bleu-gris) de la sclérotique des yeux et des cartilages des oreilles et du nez, souvent appelée ochronose.[ 10 ]
Au fil du temps, d’autres symptômes apparaissent:
- pigmentation sévère de la peau sur les pommettes, les aisselles et les parties génitales;
- coloration des vêtements lors du contact avec les zones transpirantes du corps;
- attaques de faiblesse générale;
- voix rauque.
Il convient de garder à l’esprit que l’alcaptonurie et l’ochronose, comme indiqué ci-dessus, sont des noms synonymes pour le même trouble du catabolisme de la tyrosine.
Maladie des urines à odeur de sirop d'érable et alcaptonurie. La leucinose congénitale des urines à odeur de sirop d'érable est également un trouble métabolique. Elle présente le même mode de transmission. Des mutations se produisent sur le même chromosome, mais affectent le gène codant pour le complexe enzymatique de l'α-cétoacide déshydrogénase à chaîne ramifiée. De ce fait, l'organisme ne peut pas dégrader certains composants des protéines, notamment les acides aminés leucine, isoleucine et valine. Dans ce cas, l'urine (et le cérumen) ont une odeur sucrée; de plus, le tableau clinique de ce type d'acidémie organique comprend une hypopigmentation, des fluctuations de la tension artérielle, des convulsions, des vomissements et des diarrhées, une baisse de la glycémie, une acidocétose, des hallucinations, etc. Le taux de mortalité chez les enfants est assez élevé; chez les adultes, sans traitement, le coma et le décès peuvent survenir par œdème cérébral.
L'albinisme et l'alcaptonurie ne sont liés que par la tyrosine. L'albinisme, y compris l'alcaptonurie oculocutanée, est causé par des mutations génétiques affectant la production de mélanine. Des modifications congénitales sont observées dans le gène TYR, situé sur le chromosome 11 (11q14.3), qui code pour la tyrosinase, une enzyme mélanosome contenant du cuivre et nécessaire à la formation du pigment cutané à partir des produits du métabolisme de la tyrosine. Cette maladie est beaucoup plus fréquente que l'alcaptonurie.
Complications et conséquences
Causées par l'action des métabolites intermédiaires de la tyrosine – acides homogentisique et benzoquinoneacétique – les conséquences et complications de l'alcaptonurie apparaissent en raison du dépôt de polymères pigmentés réactifs, de la destruction des fibrilles de collagène et de la détérioration de l'état du cartilage (avec une diminution de leur résistance aux contraintes mécaniques).
Au fil des années, à l'âge adulte, l'arthrite dégénérative et l'arthrose des grosses articulations (hanche, sacro-iliaque et genou) se développent; les espaces intervertébraux se rétrécissent (en particulier dans la colonne lombaire et thoracique) - avec calcification et formation d'ostéophytes; la densité du tissu des plaques osseuses sous-chondrales diminue et les os sous-jacents peuvent subir un remodelage pathologique avec formation d'excroissances et de déformations. [ 11 ]
Des lésions des valves cardiaques (aortiques et mitrales) et des artères coronaires peuvent être observées – avec des signes de maladie coronarienne, ainsi que la formation de calculs dans les reins et la prostate – en raison de la même calcification. [ 12 ], [ 13 ]
Diagnostics alcaptonurie
En règle générale, le diagnostic des troubles métaboliques congénitaux repose sur l’étude des fluides biologiques du corps.
Sur quels tests et réactions l'alcaptonurie peut-elle être diagnostiquée? Des analyses d'urine sont nécessaires pour détecter l'acide homogentisique et déterminer son taux (normal: 20 à 30 mg par jour, élevé: 3 à 8 g). Un échantillon d'urine est analysé par chromatographie en phase gazeuse ou spectrométrie de masse, en utilisant la chromatographie liquide; un test de dépistage de la présence de chlorure de fer dans les urines est possible. [ 14 ]
Il existe également une méthode de diagnostic rapide: la détermination de l’alcapton dans des taches d’urine séchées sur papier (par intensité de couleur).
Lors de la clarification du diagnostic, le diagnostic instrumental (radiographie) consiste à identifier les signes radiologiques d'arthrose et d'autres pathologies articulaires chez les patients.
Le diagnostic est confirmé par des méthodes de génétique moléculaire de diagnostic des maladies héréditaires, telles que les tests génétiques et le séquençage de l'ADN. [ 15 ]
Diagnostic différentiel
Le diagnostic différentiel comprend l'hémochromatose et l'insuffisance hépatique aiguë du nouveau-né, la mélaninurie, la porphyrie aiguë intermittente, la lymphohistiocytose hémophagocytaire, la pathologie mitochondriale primaire, la polyarthrite rhumatoïde et la spondylarthrite ankylosante.
Qui contacter?
Traitement alcaptonurie
Le principal traitement de l'alcaptonurie est l'administration orale de fortes doses (au moins 1 000 mg par jour) d'acide ascorbique. Chez l'enfant, cela augmente l'excrétion urinaire d'acide homogentisique et, chez l'adulte, réduit la teneur urinaire de son dérivé, l'acide benzoquinoneacétique, et ralentit sa liaison aux structures conjonctives des articulations et au collagène. [ 16 ]
Des cliniques d'Europe occidentale testent actuellement la nitisinone (Orfalin), un médicament appartenant au groupe des métabolites qui inhibe la deuxième étape du catabolisme de la tyrosine: la transformation du para-hydroxyphénylpyruvate en acide homogentisique. Cependant, l'utilisation de cet agent pharmacologique entraîne une accumulation de tyrosine et peut entraîner des effets secondaires graves, notamment une opacité cornéenne et une photophobie, des saignements de nez et d'estomac, une insuffisance hépatique, des modifications sanguines, etc. Néanmoins, aux États-Unis, la nitisinone est approuvée par la FDA pour le traitement de la tyrosinémie de type I. [17 ], [ 18 ]
Ainsi, des traitements de physiothérapie – thérapie par l’exercice pour augmenter la force musculaire et améliorer la mobilité articulaire, balnéothérapie et thérapie péloïde pour limiter la douleur – sont réalisés pour les problèmes articulaires causés par l’alcaptonurie.
Bien que la tyrosine ne soit pas seulement fournie par l'alimentation mais également produite par l'organisme, il est recommandé aux patients atteints d'alcaptonurie de suivre un régime pauvre en protéines et de limiter la consommation d'aliments riches en tyrosine, principalement le bœuf et le porc, les produits laitiers (en particulier les fromages), les légumineuses, les noix et les graines.
La prévention
La prévention des mutations génétiques est impossible, mais pour éviter la naissance d'enfants présentant un risque élevé de troubles congénitaux, il existe un conseil génétique médical, qui est nécessaire avant une grossesse planifiée pour les couples dont les antécédents familiaux comprennent des maladies héréditaires. [ 19 ]
Prévoir
Les issues fatales de l'alcaptonurie sont très rares, et le décès peut être dû à de graves complications cardiaques et rénales. L'espérance de vie globale des personnes atteintes d'alcaptonurie est donc bonne.
Mais la qualité de vie est réduite en raison de douleurs intenses au niveau des articulations ou de la colonne vertébrale avec une limitation importante de la mobilité, souvent progressive.