Expert médical de l'article

Nouvelles publications
Polymyosite et dermatomyosite: causes, symptômes, diagnostic, traitement
Dernière revue: 23.04.2024

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
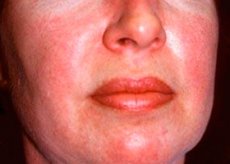
La polymyosite et la dermatomyosite sont des maladies rhumatismales systémiques rares caractérisées par des modifications musculaires inflammatoires et dégénératives (polymyosite) ou des muscles et de la peau (dermatomyosite). La manifestation cutanée la plus spécifique est une éruption d'héliotrope.
Les lésions musculaires sont symétriques et comprennent une faiblesse, une douleur et une atrophie subséquente des muscles proximaux de la ceinture des membres supérieurs. Les complications peuvent inclure des dommages aux organes internes et la malignité. Le diagnostic est basé sur l'analyse du tableau clinique et l'évaluation des troubles musculaires en déterminant les concentrations des enzymes pertinentes, en effectuant l'IRM, l'électromyographie et la biopsie du tissu musculaire. Le traitement utilise des glucocorticoïdes, parfois en association avec des immunosuppresseurs ou des immunoglobulines administrées par voie intraveineuse.
Les femmes sont malades deux fois plus souvent que les hommes. La maladie peut survenir à tout âge, mais est plus souvent détectée dans l'intervalle de 40 à 60 ans; chez les enfants de 5 à 15 ans.
 [
[Quelles sont les causes de la dermatomyosite et de la polymyosite?
La cause de la maladie est censée être une réaction auto-immune au tissu musculaire chez les individus génétiquement prédisposés. La maladie est plus fréquente en présence d'antécédents familiaux accablés et porteurs de certains antigènes HLA (DR3, DR52, DR56). Les facteurs de démarrage possibles sont la myosite virale et les néoplasmes malins. Il y a des rapports de la détection dans les cellules musculaires de structures semblables aux picornavirus; En outre, les virus peuvent induire des maladies similaires chez les animaux. Association de tumeurs malignes avec la dermatomyosite (beaucoup moins avec la polymyosite) suggère que la croissance tumorale peut également être un mécanisme de déclenchement de maladies résultant de l' exécution de l' auto - immunité à des antigènes de tumeur partagés et le tissu musculaire.
Dans les parois des vaisseaux sanguins des muscles squelettiques, des dépôts d'IgM, d'IgG et du troisième composant du complément sont détectés; ceci est particulièrement vrai chez les enfants atteints de dermatomyosite. Les patients atteints de polymyosite peuvent également développer d'autres processus auto-immuns.
Physiopathologie dermatomyosite et polymyosite
Les changements pathologiques comprennent les dommages cellulaires et leur atrophie sur fond d'inflammation de sévérité variable. Les muscles des membres supérieurs et inférieurs, ainsi que le visage, sont moins affectés que les autres muscles squelettiques. La défaite de la musculature viscérale du pharynx et des parties supérieures de l'œsophage, moins souvent du cœur, de l'estomac ou de l'intestin, peut entraîner une perturbation des fonctions de ces organes. Des concentrations élevées de myoglobine, causées par la rhabdomyolyse, peuvent causer des dommages aux reins. Il peut aussi y avoir des changements inflammatoires dans les articulations et les poumons, en particulier chez les patients qui ont des anticorps antisynthétiques.
Les symptômes de la dermatomyosite et polymyosite
L'apparition de la polymyosite peut être aiguë (surtout chez les enfants) ou subaiguë (habituellement chez les adultes). L'infection virale aiguë précède parfois ou est le facteur de départ de la manifestation de la maladie, dont les manifestations les plus fréquentes sont la faiblesse des muscles proximaux ou les éruptions cutanées. Les sensations de la douleur sont moins exprimées que la faiblesse. Peut-être le développement de la polyarthralgie, le phénomène de Raynaud, la dysphagie, les violations des poumons, les symptômes communs (fièvre, diminution de sa masse, faiblesse). Le phénomène de Reynaud est souvent retrouvé chez les patients ayant des maladies conjonctives concomitantes.
La faiblesse musculaire peut progresser pendant plusieurs semaines ou plusieurs mois. Cependant, pour la manifestation clinique de la faiblesse musculaire, un minimum de 50% des fibres musculaires doit être affecté (ainsi, la présence d'une faiblesse musculaire indique une progression de la myosite). Les patients peuvent avoir de la difficulté à lever les bras au-dessus du niveau des épaules, à monter les escaliers et à se lever de la position assise. En raison de la faiblesse prononcée des muscles pelviens et de la ceinture scapulaire, les patients peuvent être rivés à un fauteuil roulant ou à un lit. Avec la défaite du fléchisseur du cou, il devient impossible de déchirer la tête de l'oreiller. La défaite des muscles du pharynx et des parties supérieures de l'œsophage entraîne une violation de la déglutition et de la régurgitation. Les muscles des membres inférieurs et supérieurs et du visage ne sont généralement pas affectés. Cependant, le développement de contractures des membres est possible.
Les éruptions cutanées, notées avec la dermatomyosite, ont généralement une couleur foncée et un caractère érythémateux. Gonflement périorbitaire de couleur pourpre (éruption héliotrope) est également caractéristique. Les éruptions cutanées peuvent légèrement dépasser le niveau de la peau et être lisses ou couvertes d'écailles; la localisation des lésions - le front, le cou, les épaules, la poitrine, le dos, les avant-bras, les parties inférieures des sourcils des cuisses, des genoux, de la malléole interne, la surface arrière des articulations interphalangiennes et métacarpophalangiennes, avec le côté latéral (symptômes Gottrona). Hyperémie possible de la base ou de la périphérie des ongles. Sur la peau de la surface latérale des doigts, il est possible de développer une dermatite desquamative, accompagnée de l'apparition de fissures. Lésions cutanées primaires souvent résolues sans conséquences, mais peut conduire au développement de changements secondaires sous forme de pigmentation foncée, atrophie, cicatrices ou vitiligo. Possible la formation de calcifications sous-cutanées, en particulier chez les enfants.
Environ 30% des patients développent une polyarthralgie ou une polyarthrite, souvent accompagnée d'un gonflement et d'un épanchement articulaire. Néanmoins, la sévérité des manifestations articulaires est faible. Plus souvent, ils se produisent lorsque les patients ont des anticorps contre Jo-1 ou d'autres synthétases.
La défaite des organes internes (à l'exception du pharynx et de l'œsophage supérieur) dans la polymyosite est moins fréquente que dans d'autres maladies rhumatismales (en particulier le LES et la sclérodermie systémique). Rarement, surtout avec un syndrome antisynthétique, la maladie se manifeste par une pneumonie interstitielle (sous forme de dyspnée et de toux). Des arythmies cardiaques et des troubles de la conduction peuvent se développer, mais ils sont généralement asymptomatiques. Les manifestations du tractus gastro-intestinal sont plus fréquentes chez les enfants qui souffrent également de vascularite, et peuvent inclure des vomissements avec un mélange de sang, de méléna et de perforation de l'intestin.
Où est-ce que ça fait mal?
Classification de la polymyosite
Il y a 5 variantes de polymyosite.
- Polymyosite idiopathique primaire, qui peut survenir à tout âge. Avec elle, il n'y a pas de lésion cutanée.
- La dermatomyosite idiopathique primaire est semblable à la polymyosite idiopathique primaire, mais avec elle il y a une lésion de la peau.
- Une polymyosite et une dermatomyosite associées à des néoplasmes malins peuvent survenir chez des patients de tout âge; le plus souvent observé chez les patients âgés, ainsi que chez les patients souffrant d'autres maladies du tissu conjonctif. Le développement de néoplasmes malins peut être observé à la fois dans les 2 ans avant et dans les 2 ans après le début de la myosite.
- La polymyosite infantile ou dermatomyosite est associée à une vascularite systémique.
- La polymyosite et la dermatomyosite peuvent également survenir chez des patients souffrant d'autres maladies du tissu conjonctif, le plus souvent avec une sclérose systémique progressive, une maladie mixte du tissu conjonctif et un LED.
L'inclusion du muscle du tronc dans le groupe de polymyosite de la myosite est incorrecte, car cette dernière est une maladie distincte caractérisée par des manifestations cliniques similaires à celles d'une polymyosite idiopathique chronique. Cependant, il se développe dans le grand âge, affecte souvent les muscles des parties distales du corps (par exemple, les extrémités supérieures et inférieures), a une durée plus longue, répond mal au traitement et est caractérisé par une image histologique typique.
Le diagnostic de la dermatomyosite et la polymyosite
La poliomyosite devrait être suspectée chez les patients présentant des plaintes de faiblesse des muscles proximaux, accompagnés de leur douleur ou sans elle. Test de la dermatomyosite est nécessaire pour les patients se plaignent d'une éruption cutanée ressemblant à héliotrope, ou un symptôme Gottrona, ainsi que chez les patients présentant des manifestations de la polymyosite, associée à des lésions de la peau, la dermatomyosite appropriée. Les manifestations cliniques de la polymyosite et la dermatomyosite peuvent ressembler à ceux de la sclérose systémique ou, moins fréquemment - SLE ou vascularite. La fiabilité du diagnostic est accrue en faisant correspondre le plus grand nombre possible de ces cinq critères:
- faiblesse des muscles proximaux;
- éruptions cutanées caractéristiques;
- activité accrue des enzymes du tissu musculaire (créatine kinase ou, en l'absence d'augmentation de son activité, aminotransférases ou aldolase);
- changements caractéristiques dans la myographie ou l'IRM;
- changements histologiques caractéristiques dans la biopsie du tissu musculaire (critère absolu).
Une biopsie musculaire peut exclure certaines conditions cliniquement similaires, telles que le muscle du tronc musculaire et la rhabdomyolyse causée par une infection virale. Les changements révélés par l'examen histologique peuvent être différents, mais typiques sont l'inflammation chronique, les foyers de dégénérescence et la régénération des muscles. Avant le début d'un traitement potentiellement toxique, un diagnostic précis doit être établi (généralement par une vérification histologique). Avec l'IRM, il est possible d'identifier les foyers d'œdème et d'inflammation des muscles suivis d'une biopsie ciblée.
Des études en laboratoire permettent de renforcer ou, au contraire, pour éliminer la suspicion de la présence de la maladie, et sont également utiles pour évaluer sa gravité, combinaison possible avec d'autres diagnostics et pathologie complications similaires. Malgré le fait que des anticorps antinucléaires soient détectés chez certains patients, ce phénomène est plus typique pour d'autres maladies du tissu conjonctif. Environ 60% des patients ont des anticorps contre l'antigène nucléaire (PM-1) ou des cellules entières du thymus et Jo-1. Le rôle des auto-anticorps dans la pathogenèse de la maladie reste peu claire, mais on sait que des anticorps contre Jo-1 est marqueur spécifique antisintetaznogo syndrome comprenant l'alvéolite fibrosante, la fibrose pulmonaire, l'arthrite, le phénomène de Raynaud.
L'évaluation périodique de l'activité de la créatine kinase est utile pour surveiller le traitement. Néanmoins, avec une atrophie musculaire sévère, l'activité de l'enzyme peut être normale, malgré la présence d'une myosite active chronique. Les données d'IRM, les biopsies musculaires ou les valeurs élevées de l'activité de la créatine kinase aident souvent à différencier la récurrence de la polymyosite et de la myopathie induite par les glucocorticoïdes.
Parce que de nombreux patients ont un cancer non diagnostiqué, certains auteurs recommandent un dépistage tous les adultes avec dermatomyosite, et les personnes souffrant de polymyosite, à l'âge de 60 comme suit: examen physique, examen des seins vkpyuchayuschy, l'examen gynécologique et un examen du rectum ( y compris l'étude des matières fécales pour le sang latent); test sanguin clinique; des tests sanguins biochimiques; mammographie; détermination de l'antigène embryonnaire cancéreux; analyse générale de l'urine; radiographie thoracique. La nécessité d'un tel dépistage pour les patients plus jeunes qui n'ont pas de signes cliniques de tumeurs malignes a été remise en question par certains auteurs.
Qu'est-ce qu'il faut examiner?
Comment examiner?
Traitement de la dermatomyosite et de la polymyosite
Avant d'arrêter l'inflammation, il est nécessaire de limiter l'activité physique. Les glucocorticoïdes sont des médicaments de première intention. Au stade aigu de la maladie, les patients adultes ont besoin de prednisolone (à l'intérieur) à une dose de 40 à 60 mg par jour. La détection régulière de l'activité de la créatine kinase est un indicateur précoce de l'efficacité: chez la plupart des patients, sa diminution ou normalisation se produit dans les 6 à 12 semaines suivant l'augmentation de la force musculaire. Après la normalisation de l'activité enzymatique, la dose de prednisolone diminue: d'abord environ 2,5 mg par jour pendant la semaine, puis plus rapidement; lorsque l'activité des enzymes musculaires augmente, la dose de l'hormone est de nouveau augmentée. Les patients guéris peuvent se passer de glucocorticoïdes, mais le plus souvent, les patients adultes ont besoin d'un traitement glucocorticoïde à long terme (10-15 mg de prednisolone par jour). La dose initiale de prednisolone pour les enfants est de 30-60 mg / m 2 une fois par jour. S'il y a une rémission> 1 an, les enfants peuvent avoir un arrêt de la thérapie glucocorticoïde.
Dans certains cas, chez les patients recevant des doses élevées de glucocorticoïdes, une augmentation soudaine de la faiblesse musculaire se produit, ce qui peut être dû au développement d'une myopathie glucocorticoïde.
Lorsque une réponse inadéquate au traitement glucocorticoïde et aussi dans le développement de la myopathie glucocorticoïde ou d'autres complications nécessitant une réduction de la dose ou prednisolone de retrait, utilisez immunodépresseurs (méthotrexate, cyclophosphamide, azathioprine, ciclosporine). Certains patients ne peuvent recevoir le méthotrexate (habituellement à des doses plus élevées que celles dans le traitement de la polyarthrite rhumatoïde) pendant plus de 5 ans. Les immunoglobulines intraveineuses peuvent être efficaces chez les patients réfractaires au traitement médicamenteux, mais leur utilisation augmente le coût du traitement.
La myosite associée aux tumeurs primaires et métastatiques, ainsi que la myosite des muscles du tronc, sont généralement plus réfractaires à la thérapie par les glucocorticoïdes. Le développement de la rémission associée à des tumeurs malignes de la myosite est possible après l'ablation de la tumeur.
Quel est le pronostic de la dermatomyosite et de la polymyosite?
La rémission à long terme (et même la récupération clinique) pendant 5 ans sont notées dans plus de la moitié des patients traités; chez les enfants, cet indicateur est plus élevé. La rechute peut cependant se développer à tout moment. Le taux global de survie à cinq ans est de 75%, plus élevé chez les enfants. Les causes de décès chez les adultes sont une faiblesse musculaire sévère et progressive, une dysphagie, une diminution de la nutrition, une pneumonie par aspiration ou une insuffisance respiratoire due à des infections pulmonaires. La poliomyosite est plus sévère et plus résistante au traitement en présence de lésions du cœur et des poumons. La mort chez les enfants peut se produire en raison de la vascularite intestinale. Le pronostic global de la maladie est également déterminé par la présence de néoplasmes malins.