Expert médical de l'article

Nouvelles publications
Xeroderma pigmentosum
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
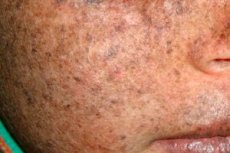
Le xeroderma pigmentosum est une maladie génétique autosomique récessive de la réparation de l'ADN. La maladie est causée par des mutations de gènes impliqués dans la réparation de l'ADN endommagé par les rayons ultraviolets et d'autres substances toxiques.
Cette maladie touche le plus souvent les enfants, aussi appelés « enfants de la nuit ». Les complications fréquentes sont le carcinome basocellulaire et d'autres néoplasmes malins de la peau, le mélanome malin métastatique et le carcinome épidermoïde.
 [ 1 ]
[ 1 ]
Causes xérodermie pigmentaire
Pathogénèse
Le déficit en endonucléases UV dans les cellules du patient (fibroblastes), voire leur absence totale, joue un rôle important dans le développement de la maladie. Cette enzyme est responsable de la reproduction de l'ADN endommagé par les rayons ultraviolets. Selon d'autres sources, le déficit en ADN polymérase-1 joue un rôle important dans le développement de la maladie chez certains patients. La maladie se développe le plus souvent sous l'influence de rayons d'une longueur d'onde de 280 à 310 nm. On pense que la xérodermie pigmentaire se développe en raison de l'entrée de diverses substances photodynamiques dans l'organisme ou d'une augmentation des porphyrines dans l'environnement biologique humain.
Aux premiers stades de la maladie, on observe une hyperkératose, une atrophie du foyer épidermique, un amincissement de la couche de Malpighi, une augmentation des granules de mélanine dans la couche basale des cellules et une inflammation chronique infiltrant la couche supérieure du derme (principalement autour des vaisseaux sanguins). Par la suite, des modifications dégénératives du collagène et des fibres élastiques sont détectées, et au stade tumoral, des signes caractéristiques d'un cancer de la peau sont révélés.
Symptômes xérodermie pigmentaire
Hommes et femmes sont autant touchés par cette maladie. Elle débute dès la petite enfance, au printemps ou en été. La peau du patient est particulièrement sensible au soleil. Par conséquent, une première éruption cutanée, sous forme d'hyperpigmentation, semblable à un grain de beauté, apparaît sur les zones cutanées exposées au soleil. L'éruption s'aggrave, l'érythème s'intensifie, devient brun foncé, des télangiectasies, un angiome et une atrophie apparaissent. Par la suite, des papillomes et des verrues se développent (principalement sur la peau du visage et du cou), des taches et des ulcères apparaissent. Suite aux cicatrices des ulcères, le nez s'amincit (« en bec d'oiseau ») et la paupière s'éverse. Les papillomes évoluent généralement vers des tumeurs malignes. La xérodermie pigmentée accompagne généralement un cancer spinocellulaire ou un mélanosarcome.
Qu'est ce qui te tracasse?
Étapes
Dans l'évolution clinique du xeroderma pigmentosum, on distingue cinq stades: inflammatoire (érythémateux), hyperpigmentation, atrophie, hyperkératose et tumeurs malignes.
Au stade inflammatoire (érythémateux), des gonflements, des taches rouges et parfois des cloques et des vésicules apparaissent sur les zones cutanées exposées au soleil (visage, cou, haut du thorax, bras, mains). L'éruption cutanée est quasiment absente sur les zones non exposées au soleil.
En cas d'hyperpigmentation, des taches hyperpigmentées brun clair, semblables à des grains de beauté, apparaissent à la place des taches rouges.
Avec l'atrophie, la peau se dessèche, s'amincit et se ride. De nombreuses petites télangiectasies étoilées et cicatrices à surface brillante sont observées sur la peau des lèvres et du nez. L'atrophie et les cicatrices entraînent une microtomie (réduction de l'ouverture buccale), un ectropion, un amincissement des oreilles et du nez, ainsi qu'une atrésie de l'ouverture nasale et de la bouche. Chez 80 à 85 % des patients, les yeux sont touchés: conjonctivite, kératite, lésions de la cornée et des muqueuses, et baisse de la vision. Une dyschromie, des télangiectasies, des lésions hyperkératosiques et des tumeurs sont observées sur la peau des paupières.
L'hyperkératose peut entraîner l'apparition de tumeurs verruqueuses, de papillomes, de kératoacanthomes, de fibromes, d'angiofibriomes et d'autres tumeurs bénignes dans les foyers pathologiques décrits ci-dessus. C'est pourquoi certains scientifiques classent la xérodermie pigmentaire parmi les maladies précancéreuses.
Dix à quinze ans après le début de la maladie, des tumeurs cutanées malignes (basaliome, endothéliome, angiosarcome) apparaissent sous forme de foyers atrophiés et de taches pigmentaires. Ces tumeurs sont rapidement détruites, métastasent aux organes internes et entraînent le décès. Chez certains patients, des modifications dystrophiques générales des organes et tissus internes sont observées (syndactélie des deuxième et troisième orteils, dystrophie dentaire, perte complète des cheveux, etc.).
Formes
La forme neurologique du xeroderma pigmentosum se manifeste par deux syndromes.
Le syndrome de Reed se caractérise par des manifestations cliniques de xeroderma pigmentosum et de microcéphalie, une idiopathie et un ralentissement de la croissance du système squelettique. Dans la forme neurologique de la maladie, la réparation de l'ADN est difficile et la radiothérapie entraîne une augmentation des principaux foyers pathologiques.
Le syndrome de De Sinctis-X Cocchione est caractérisé par les signes cliniques suivants:
- développement de signes cliniques de xeroderma pigmentosum sur des peaux particulièrement photosensibles;
- apparition précoce de tumeurs malignes;
- évolution simultanée de paralysie spastique, de microcéphalie et de démence;
- malformations congénitales et nanisme;
- sous-développement des gonades;
- fausses couches fréquentes;
- transmission récessive par hérédité.
Selon certains dermatologues, le syndrome de Santis-Cacchione n'est pas une maladie indépendante, mais une manifestation clinique grave et pleinement exprimée du xeroderma pigmentosum.
Formes génétiques
Types |
Gène |
Lieu |
Description |
Type A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) groupe A – forme classique |
Type B, II, XPB |
XPB |
2q21 |
XP Groupe B |
Type C, III, XPC |
XPC |
3p25 |
XP Groupe C |
Type D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP Groupe D ou Syndrome De Sanctis-Cacchione |
Type E, V, XPE |
DDB2 |
11p12-p11 |
XP Groupe E |
Type F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Groupe F |
Type G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP groupe G et syndrome COFS (syndrome cérébro-oculo-facio-squelettique) de type 3 |
Type V, XPV |
POLH |
6p21.1-p12 |
Variante de Xeroderma Pigmentosa |
Qu'est-ce qu'il faut examiner?
Comment examiner?
Qui contacter?
Traitement xérodermie pigmentaire
Les antipaludiques (délagyl, plaquenil, résoquine, etc.), qui protègent l'ADN des rayons ultraviolets, inhibent le processus de dépolymérisation et réduisent la sensibilité (photosensibilisation) de la peau au soleil. Ces médicaments ont des propriétés anti-inflammatoires et hyposensibilisantes. Il est recommandé d'associer un traitement général à une vitaminothérapie (B1, B2, PP, B6, B12, A, E), des antihistaminiques (tavegil, diphenhydramine, suprastine) et des agents désensibilisants (thiosulfate de sodium, chlorure de calcium à 10 % par voie intraveineuse (10 ml).
Pour le traitement local, des crèmes et des onguents solaires sont utilisés.
Dans la forme tumorale de xérodermie pigmentaire, des méthodes chirurgicales, l'azote liquide et les rayons laser sont utilisés. Pour vous protéger du soleil, il est nécessaire de porter des vêtements amples, un chapeau et des gants.
Prévoir
La plupart des patients (2/3) décèdent avant d’atteindre l’âge de 15 ans. Certains patients peuvent vivre jusqu’à 40-50 ans.
[ 20 ]