Expert médical de l'article

Nouvelles publications
Syndrome d'Usher
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Le syndrome d'Usher est une maladie héréditaire qui se manifeste par une surdité totale dès la naissance, ainsi que par une cécité progressive avec l'âge. La perte de vision est associée à une rétinite pigmentaire, une dégénérescence pigmentaire de la rétine. De nombreuses personnes atteintes du syndrome d'Usher présentent également de graves troubles de l'équilibre.
Épidémiologie
Grâce à cette recherche, il a été possible d'établir que le syndrome d'Usher touche environ 8 % des enfants sourds-muets examinés (les tests ont été réalisés dans des établissements spécialisés pour sourds-muets). Une rétinite pigmentaire a été observée chez 6 à 10 % des patients atteints de surdité congénitale, elle-même observée chez environ 30 % des personnes atteintes d'une maladie de la rétine pigmentaire.
On estime que cette maladie touche environ 3 à 10 personnes sur 100 000 dans le monde. Elle touche aussi bien les femmes que les hommes. Environ 5 à 6 % de la population mondiale souffre de ce syndrome. Environ 10 % des cas de surdité profonde infantile sont dus au syndrome d'Usher de type I ou II.
Aux États-Unis, les types 1 et 2 sont les plus courants. Ensemble, ils représentent environ 90 à 95 % des cas de syndrome d'Usher chez l'enfant.
Causes Syndrome d'Usher
Les syndromes d'Usher de types I, II et III ont une origine autosomique récessive, tandis que le type IV est considéré comme une anomalie du chromosome X. Les causes de cécité et de surdité associées à ce syndrome n'ont pas encore été suffisamment étudiées. On suppose que les personnes atteintes de cette maladie présentent une hypersensibilité à des composants susceptibles d'endommager la structure de l'ADN. De plus, cette maladie pourrait être associée à des troubles du système immunitaire, mais dans ce cas, il n'existe pas de description précise de ce processus.
En 1989, des anomalies chromosomiques ont été identifiées pour la première fois chez des patients atteints de la maladie de type II, ce qui pourrait permettre à l'avenir d'isoler les gènes responsables du syndrome. Il pourrait également être possible d'identifier ces gènes chez les porteurs et de développer des tests génétiques prénataux spécifiques.
 [ 8 ]
[ 8 ]
Facteurs de risque
Le syndrome est transmissible lorsque les deux parents sont atteints, c'est-à-dire selon un mode récessif. Un enfant peut également hériter de la maladie si ses parents sont porteurs du gène. Si les deux futurs parents sont porteurs de ce gène, la probabilité d'avoir un enfant atteint de ce syndrome est de 1 sur 4. Une personne ne possédant qu'un seul gène du syndrome est considérée comme porteuse, mais ne présente aucun symptôme de la maladie. À l'heure actuelle, il n'est pas encore possible de déterminer si une personne est porteuse du gène de cette maladie.
Si un enfant naît de parents dont l'un n'a pas un tel gène, la probabilité qu'il hérite du syndrome est très faible, mais il sera certainement porteur.
Symptômes Syndrome d'Usher
Les symptômes du syndrome d'Usher comprennent une perte auditive et une accumulation anormale de cellules pigmentées dans les structures oculaires. Le patient développe ensuite une dégénérescence de la rétine, entraînant une détérioration de la vision, voire une perte de vision dans les cas les plus graves.
La perte auditive neurosensorielle peut être légère ou complète et ne progresse généralement pas dès la naissance. Cependant, une maladie pigmentaire de la rétine peut se développer dès l'enfance ou plus tard. Les résultats des tests ont montré que l'acuité visuelle centrale peut être maintenue pendant de nombreuses années, même en cas de détérioration de la vision périphérique (une affection appelée « vision tunnel »).
Ce sont les principales manifestations de la maladie, qui peuvent parfois être complétées par d’autres troubles, tels que la psychose et d’autres troubles mentaux, des problèmes de l’oreille interne et/ou la cataracte.
Formes
Au cours de la recherche, 3 types de cette maladie ont été identifiés, ainsi qu'une 4ème forme, qui est assez rare.
Le type I de la maladie se caractérise par une surdité congénitale complète et des troubles de l'équilibre. Souvent, ces enfants ne commencent à marcher qu'à l'âge d'un an et demi. La détérioration de la vision débute généralement vers 10 ans, et la cécité nocturne se développe définitivement vers 20 ans. Les enfants atteints de ce type de maladie peuvent développer une détérioration progressive de la vision périphérique.
Dans la maladie de type II, on observe une surdité modérée ou congénitale. Dans ce cas, la surdité partielle ne s'aggrave souvent plus. La rétinite pigmentaire commence à se développer vers la fin de l'adolescence ou après 20 ans. La cécité nocturne apparaît généralement entre 29 et 31 ans. La baisse d'acuité visuelle dans la pathologie de type II progresse généralement un peu plus lentement que dans la pathologie de type I.
Le type III de la maladie se caractérise par une perte auditive progressive, débutant généralement pendant la puberté, ainsi que par le développement progressif au cours de la même période (légèrement plus tard que la perte auditive) d'une rétinite pigmentaire, qui peut devenir un facteur dans le développement d'une cécité progressive.
Les manifestations de la pathologie de type IV touchent principalement les hommes. Dans ce cas, des troubles progressifs et une perte de l'audition et de la vision sont également observés. Cette forme est très rare et est généralement d'origine chromosomique X.
Diagnostics Syndrome d'Usher
Le diagnostic du syndrome d'Usher est posé sur la base de la combinaison observée chez le patient d'une surdité soudaine et d'une perte progressive de la vision.
Tests
Un test génétique spécial peut être demandé pour détecter la mutation.
Onze loci génétiques pouvant provoquer le développement du syndrome d'Usher ont été découverts, et neuf gènes ont été identifiés comme étant définitivement la cause du trouble:
- Type 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Type 2: ush2a, VLGR1, WHRN.
- Syndrome d'Usher de type 3: USH3A.
Les scientifiques du NIDCD, en collaboration avec des collègues d'universités de New York et d'Israël, ont identifié une mutation appelée R245X dans le gène Pcdh15 qui est responsable d'un pourcentage important du syndrome d'Usher de type 1 dans la population juive.
Pour en savoir plus sur les laboratoires qui réalisent des essais cliniques, visitez https://www.genetests.org et recherchez « syndrome d'Usher » dans l'annuaire des laboratoires.
Pour en savoir plus sur les essais cliniques existants qui incluent des tests génétiques pour le syndrome d'Usher, visitez https://www.clinicaltrials.gov et recherchez « syndrome d'Usher » ou « tests génétiques du syndrome d'Usher ».
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Diagnostic instrumental
Il existe plusieurs méthodes de diagnostic instrumental:
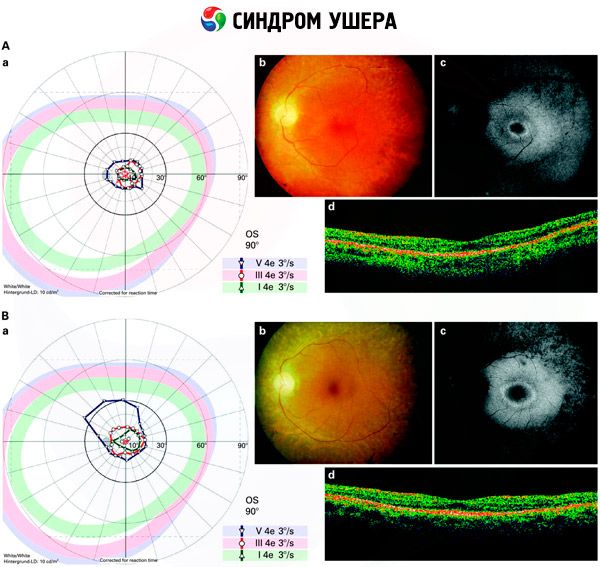
- Examen du fond d'œil pour détecter la présence de taches pigmentaires sur la rétine, ainsi qu'un rétrécissement des vaisseaux rétiniens;
- L'électrorétinogramme permet de détecter les anomalies dégénératives initiales de la rétine. Il met en évidence l'extinction des voies électroradiographiques.
- Un électronystagmogramme (ENG) mesure les mouvements oculaires involontaires qui pourraient indiquer la présence d'un déséquilibre.
- L'audiométrie, qui permet de déterminer la présence de surdité et sa gravité.
Diagnostic différentiel
Le syndrome d’Usher doit être différencié de certains troubles similaires.
Le syndrome de Hallgren se caractérise par une perte auditive congénitale et une perte progressive de la vision (cataracte et nystagmus se développent également). Les symptômes incluent également une ataxie, des troubles psychomoteurs, une psychose et un retard mental.
Le syndrome d'Alström est une maladie héréditaire caractérisée par une dégénérescence de la rétine, entraînant une perte de la vision centrale. Ce syndrome est associé à l'obésité infantile. Parallèlement, le diabète sucré et la perte auditive commencent à se développer après 10 ans.
Chez une femme enceinte, la rubéole au premier trimestre peut entraîner diverses anomalies du développement de l'enfant. Parmi les conséquences de ces anomalies figurent une perte auditive, des troubles de la vue et divers troubles du développement.
Qui contacter?
Traitement Syndrome d'Usher
Il n'existe actuellement aucun traitement curatif contre le syndrome d'Usher. Par conséquent, le traitement consiste principalement à ralentir la perte de vision et à compenser la perte auditive. Les traitements possibles incluent:
- Prendre de la vitamine A (certains ophtalmologistes pensent que des doses élevées de palmitate de vitamine A peuvent ralentir, mais pas arrêter, la progression de la rétinite pigmentaire);
- Implantation d'appareils électroniques spéciaux dans les oreilles du patient (appareils auditifs, implants cochléaires).
Les ophtalmologistes recommandent à la plupart des adultes atteints de formes courantes de rétinite pigmentaire de prendre 15 000 UI (unités internationales) de palmitate de vitamine A par jour, sous surveillance. Les personnes atteintes du syndrome d'Usher de type 1 n'ayant pas été incluses dans l'étude, des doses élevées de vitamine A ne sont pas recommandées pour ce groupe de patients. Les personnes envisageant un traitement à base de vitamine A doivent en discuter avec leur médecin. Autres recommandations:
- Modifiez votre régime alimentaire pour inclure des aliments riches en vitamine A.
- Les femmes qui envisagent de devenir enceintes devraient arrêter de prendre des doses élevées de vitamine A trois mois avant de concevoir en raison d’un risque accru de malformations congénitales.
- Les femmes enceintes devraient arrêter de prendre des doses élevées de vitamine A en raison d’un risque accru de malformations congénitales.
Il est également important d'adapter cet enfant à la vie sociale. Cela nécessite l'aide d'éducateurs spécialisés et de psychologues. Si le patient présente une perte progressive de la vision, il convient de lui apprendre à utiliser la langue des signes.
Prévoir
Le pronostic du syndrome d'Usher est défavorable. Chez la plupart des patients atteints de cette maladie, quel que soit son type, le champ visuel et son acuité visuelle commencent à se détériorer vers 20 à 30 ans. Dans certains cas, une perte totale de la vision bilatérale survient. La perte auditive, toujours accompagnée de mutisme, évolue très rapidement vers une perte auditive bilatérale complète.