Expert médical de l'article

Nouvelles publications
Steatocystoma
Dernière revue: 29.06.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
Le stéatocystome (synonyme: sébocystome) est une tumeur bénigne, non mictionnelle, remplie de sécrétions graisseuses. Il est rare et les éruptions cutanées peuvent être solitaires, simples (stéatocystome simplex) ou multiples. Le stéatocystome multiple est une maladie génétique rare. [ 1 ] Il se caractérise par de multiples kystes dermiques de tailles variables, présents dans les zones où l'unité pilo-sébacée est bien développée, principalement au niveau des aisselles, du tronc et des extrémités. [ 2 ] Chez la femme, le stéatocystome est plus fréquemment observé dans la région inguinale, et chez l'homme, il a généralement une distribution en forme de losange sur le tronc. Rarement observées sur le cuir chevelu et le visage, les lésions vont de normochromes à jaunes, sont mobiles, à croissance lente et contiennent principalement du liquide.
Le stéatocystome apparaît à l'adolescence ou au début de l'âge adulte. Dans la plupart des cas, la maladie est de type autosomique dominant, mais elle peut également survenir de manière sporadique. [ 3 ], [ 4 ]
Causes ng steatocystomas
Cette pathologie dermatologique étant assez rare, ses causes d'apparition ne sont pas précisément étudiées et restent hypothétiques. Des cas familiaux de la maladie sur plusieurs générations sont connus; on suppose qu'elle est transmise selon le mode autosomique dominant. Parallèlement, la littérature médicale décrit de nombreux cas isolés (sporadiques) de stéatocystome, et de nombreux patients présentaient des kératoses congénitales ou acquises concomitantes, ainsi que d'autres maladies systémiques.
La cause présumée des stéatokystes simples ou multiples est la stimulation androgénique (progestérone) des glandes sébacées, qui survient lors des changements hormonaux de l'adolescence. Cette hypothèse repose sur le fait que la maladie apparaît dans la grande majorité des cas chez les adolescents ou les jeunes adultes après la puberté. L'influence d'un environnement défavorable, d'un traumatisme, d'une infection ou d'un dysfonctionnement du système immunitaire n'est pas exclue.
Il n'existe aucune statistique sur l'incidence de la maladie. Certains auteurs affirment que la stéatocytomatose est plus fréquente chez les jeunes hommes, tandis que d'autres estiment que le sexe n'a aucune importance. L'âge d'apparition des éruptions cutanées varie principalement entre 12 et 25 ans et elles persistent toute la vie. On observe également des cas isolés de manifestations précoces chez le nourrisson et de manifestations tardives chez les personnes âgées.
La forme familiale peut être associée à une mutation de la kératine 17, une kératine de type 1 présente dans les glandes sébacées et les follicules pileux. La même mutation est retrouvée dans la pachyonychie congénitale de type 2, à laquelle peut être associé le stéatocystome, une maladie autosomique dominante qui se manifeste par une dystrophie unguéale, une kératodermie palmaire et plantaire, une leucoplasie buccale, une kératose folliculaire et des kystes d'inclusion épidermiques. De plus, le stéatocystome peut se présenter avec un lichen plan squameux hypertrophique, une acrokératose verruciforme, une dentition natale et d'autres manifestations.
Au sein d'une même famille, des variations phénotypiques existent, comme dans le cas de notre patient, et les sous-types peuvent se chevaucher. La même mutation du gène de la kératine 17 peut se manifester dans le stéatocystome ou la pachyonychie de type 2, seule ou combinée. Plus de 11 mutations différentes ont été décrites; cependant, le phénotype résultant est indépendant du type de mutation. Il est possible que ces manifestations correspondent à des spectres de la même maladie.
Outre le stéatocystome purulent, d'autres cas rares de stéatocystome ont été rapportés, tels que le stéatocystome facial, acral, vulvaire et simple (lésion unique).
Facteurs de risque
Certains chercheurs ont observé que le stéatocystome était associé aux pathologies congénitales suivantes avec un type d'hérédité similaire:
- Pachyonychie de type Jackson-Lawler - polykératose congénitale avec lésions unguéales sévères;
- Coilonychie;
- Collagénose héréditaire - syndrome d'Ehlers-Danlos;
- Lentiginose cardiomyopathique congénitale (syndrome du Léopard);
- Dysplasie artério-hépatique;
- Trichoblastome;
- Syndrome du nævus basocellulaire;
- Cas familiaux de syringome;
- Kératoacanthome;
- Acrokératose verruciforme;
- syndrome de Gardner;
- Polykystose rénale;
- Sinus préauriculaire bilatéral.
Le risque de stéatocystome est plus élevé chez les patients atteints de dermatite de nature auto-immune présumée - hidradénite suppurée suppurée et poux plats rouges, collagénoses - polyarthrite rhumatoïde, hypothyroïdie; personnes présentant une transpiration insuffisante et des taches blanches sur les ongles (leuconychie).
Pathogénèse
La forme familiale de la maladie est associée à une mutation génétique au locus 17q12-q21. Le gène KRT17, codant pour une protéine des structures intermédiaires filamenteuses intracellulaires contenant de la kératine, des glandes sébacées, des follicules pileux et du lit de l'ongle, est altéré. Cette mutation entraîne une altération de la kératinisation des parties supérieures des follicules pileux, entraînant une déformation des canaux sébacés et la formation de formations kystiques.
De nombreux cas sporadiques de stéatocystomatose étant également connus, d'autres facteurs étiopathogéniques, autres que l'hérédité, sont évoqués. D'autres hypothèses envisagent la pathologie comme suit:
- Hamartome de l'appareil sébacé-pilaire;
- Un type de kyste dermoïde;
- Kystes séborrhéiques de rétention;
- Formations névoïdes de follicules pileux sous-développés avec glandes sébacées attachées.
La pathogénèse de la pathologie est déclenchée par des poussées hormonales, des défaillances immunitaires, des traumatismes et des infections transmises.
Symptômes ng steatocystomas
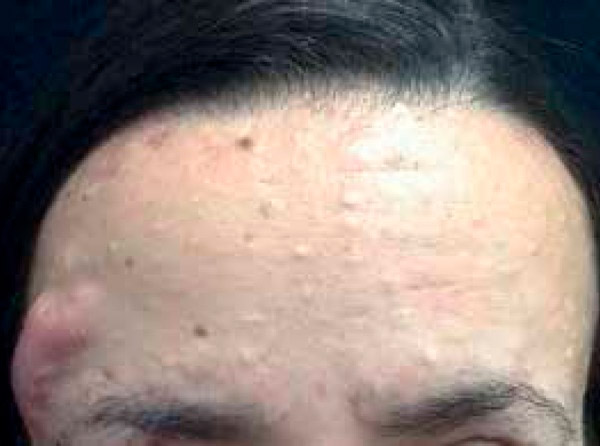
Les premiers signes sont l'apparition sur la peau d'un ou plusieurs nodules kystiques hémisphériques, mobiles, indolores à la palpation, modérément élastiques. Au début, ils sont translucides, blanchâtres ou bleutés, puis prennent une couleur jaunâtre. Le diamètre des hémisphères varie de deux à cinq millimètres. La taille du néoplasme peut parfois être plus importante; le maximum connu est de 3 cm.
Les éruptions cutanées se localisent sur les zones du corps à forte densité de structures sébacées et pileuses: l'aine, les aisselles, chez l'homme, ainsi que sur la poitrine, le scrotum, le pénis, la surface externe des mains et des pieds. Elles sont pratiquement absentes sur le cuir chevelu et le visage.
La ponction d'un stéatocystome produit un contenu huileux translucide ou une substance jaunâtre crémeuse plus épaisse dans laquelle on peut trouver des poils fins.
Selon la nature de la propagation, on distingue les formes suivantes de la maladie:
- Généralisée - propagation diffuse, impliquant le visage, le cou, le dos, la poitrine et l'abdomen, moins souvent - les extrémités, dans ces cas, modifications dystrophiques des dents et des ongles, perte de cheveux, kératinisation excessive de la peau des paumes et de la plante des pieds, troubles de la transpiration;
- Localisé - stéatocystomes multiples d'une même zone du corps;
- Solitaire ou simple - un seul néoplasme de n'importe quelle localisation.
Il existe également un sous-type purulent de stéatocystome (stéatocystome suppuré), caractérisé par une rupture spontanée des kystes suivie du développement d'une inflammation, se terminant par une cicatrisation.
Les éruptions cutanées multiples sont généralement disposées en groupes, moins souvent dispersées.
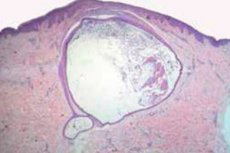
Morphologiquement, la tumeur est un kyste dont l'enveloppe est tapissée de plusieurs couches d'épithélium pavimenteux sans couche granuleuse. Sa cavité contient des poils duveteux et, sur sa face externe, des lobules de glandes sébacées.
Selon la microscopie électronique, il existe des signes de néoplasme naïf relié à la couche externe de la peau par un mince canal, constitué de cellules épithéliales indifférenciées.
Complications et conséquences
Un stéatocystome est une tumeur bénigne chronique qui présente davantage un défaut esthétique. Bien qu'il s'agisse d'une affection bénigne et que la plupart des lésions soient asymptomatiques, il existe une variante inflammatoire. Dans ce cas, on observe une rupture spontanée du kyste, une suppuration et un écoulement malodorant, notamment en cas de colonisation bactérienne secondaire. [ 5 ] Des abcès peuvent également apparaître. Ceux-ci ressemblent aux lésions de l'acné conglobate ou de l'hidradénite suppurée. Les lésions peuvent évoluer vers une cicatrisation, provoquant un inconfort important pour le patient.
La malignité est peu probable, mais il existe des preuves qu’elle est possible.
Diagnostics ng steatocystomas
Le stéatocystome est diagnostiqué sur la base des plaintes du patient, des antécédents familiaux, du tableau clinique des modifications pathologiques de la peau et des résultats de l'examen histologique du néoplasme.
Évaluer l'état général du patient. Effectuer des analyses cliniques générales (urine, sang, biochimie): les résultats sont généralement normaux. Le médecin peut prescrire des examens spécifiques et consulter un spécialiste en cas de suspicion de pathologies systémiques (polyarthrite rhumatoïde, hypothyroïdie ou hyperthyroïdie).
Un diagnostic instrumental est réalisé: microscopie du contenu tumoral et examen de sa structure. L'examen morphologique révèle la présence d'un kyste présentant une cavité de forme arbitraire, dont l'enveloppe est constituée de deux couches, sous la surface cutanée inchangée. La couche interne est tapissée de cellules épithéliales squameuses recouvertes d'une cuticule contenant des éosinophiles; la couche granuleuse n'est pas définie. La couche externe est constituée de tissu conjonctif. Des segments de glandes sébacées sont visibles dans l'enveloppe, s'ouvrant sur le follicule pileux. Les kystes sont reliés à l'épiderme par de courts brins constitués de kératocytes indifférenciés. L'analyse en culture du contenu de la cavité kystique (en l'absence d'inflammation) ne révèle aucune croissance de flore bactérienne pathogène.
Les caractéristiques cliniques du stéatocystome peuvent ressembler à un certain nombre de maladies: kystes duveteux, kystes myxoïdes, miliums, acné conglobate, hidradénite et pseudofolliculite, retardant le diagnostic et une surveillance appropriée. Le stéatocystome purulent doit être distingué de l'acné kystique nodulaire sévère, de l'acné conglobate, du fibroadénome infecté et de la pyodermite.
Diagnostic différentiel
Le diagnostic différentiel pose certaines difficultés, notamment lorsqu'il s'agit de distinguer un sébocystome d'un kyste éruptif de poils duveteux, d'autant plus que, dans certains cas, les deux néoplasies apparaissent simultanément. Elles se différencient par des caractéristiques morphologiques. La cavité d'un kyste éruptif de poils duveteux est recouverte d'un épithélium pavimenteux (comme celle d'un sébocystome), mais une couche granuleuse est présente et de nombreux poils duveteux sont observés.
En outre, une distinction est faite avec les entités suivantes:
- Kystes épidermiques et dermoïdes;
- Kystes contenant de la kératine (miliums);
- Lipomes (communément appelés tumeurs graisseuses);
- Adénome symétrique des glandes sébacées (maladie de Pringle-Burneyville);
- Syringome;
- syndrome de Gardner;
- Calcinose de la peau;
- Transpiration apocrine;
- Acné conglobate;
- Acné kystique.
Traitement ng steatocystomas
Le traitement est varié et généralement insatisfaisant en raison de la difficulté d’accès à ces lésions disséminées.
Les stéatocystomes sont retirés principalement à des fins esthétiques. Les méthodes suivantes sont utilisées:
- Excision chirurgicale;
- Aspiration à l'aiguille;
- Ablation au couteau à ondes radioélectriques;
- Électrocoagulation;
- Cryodestruction;
- Destruction au laser.
Le meilleur effet cosmétique est obtenu par traitement par ondes radio et laser.
En cas de stéatocystome purulent, un traitement systémique par isotrétinoïne et tétracycline est utilisé, avec ouverture et drainage de la cavité kystique. L'administration intrathécale d'acétonide de triamcénolone est prescrite.
Les lésions inflammatoires peuvent être traitées par injections de corticostéroïdes ou drainage. Des résultats mitigés ont été rapportés avec l'isotrétinoïne orale pour le traitement du stéatocystome. L'isotrétinoïne ne guérit généralement pas la maladie, mais réduit la taille des lésions purulentes. Cette réponse thérapeutique reflète probablement l'effet anti-inflammatoire des rétinoïdes.
Il a été rapporté dans la littérature qu’aucune rechute n’a été constatée après un traitement systémique.