Expert médical de l'article

Nouvelles publications
Polymyosite et dermatomyosite: causes, symptômes, diagnostic, traitement
Dernière revue: 05.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
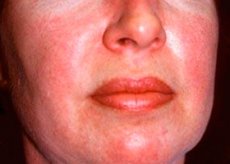
La polymyosite et la dermatomyosite sont des maladies rhumatismales systémiques rares caractérisées par des modifications inflammatoires et dégénératives des muscles (polymyosite) ou des muscles et de la peau (dermatomyosite). La manifestation cutanée la plus spécifique est l'éruption héliotrope.
L'atteinte musculaire est symétrique et se caractérise par une faiblesse, une sensibilité et une atrophie ultérieure des muscles de la ceinture pelvienne proximale. Les complications peuvent inclure une atteinte viscérale et une tumeur maligne. Le diagnostic repose sur le tableau clinique et l'évaluation du dysfonctionnement musculaire par la mesure des taux d'enzymes, l'IRM, l'électromyographie et la biopsie musculaire. Le traitement repose sur les glucocorticoïdes, parfois associés à des immunosuppresseurs ou à des immunoglobulines intraveineuses.
Les femmes sont deux fois plus souvent atteintes que les hommes. La maladie peut survenir à tout âge, mais elle est le plus souvent détectée entre 40 et 60 ans; chez les enfants, entre 5 et 15 ans.
 [
[ Quelles sont les causes de la dermatomyosite et de la polymyosite?
La cause de la maladie serait une réaction auto-immune au tissu musculaire chez des individus génétiquement prédisposés. La maladie est plus fréquente en présence d'antécédents familiaux importants et de porteurs de certains antigènes HLA (DR3, DR52, DR56). Les déclencheurs possibles sont la myosite virale et les tumeurs malignes. Des rapports ont signalé la détection de structures similaires aux picornavirus dans les cellules musculaires; de plus, des virus peuvent induire des maladies similaires chez l'animal. L'association de tumeurs malignes à la dermatomyosite (beaucoup moins fréquemment qu'à la polymyosite) suggère que la croissance tumorale pourrait également être un facteur déclenchant du développement de la maladie, résultant de l'initiation de réactions auto-immunes aux antigènes communs de la tumeur et du tissu musculaire.
Des dépôts d'IgM, d'IgG et du troisième composant du complément sont présents dans les parois des vaisseaux sanguins des muscles squelettiques; ceci est particulièrement caractéristique de la dermatomyosite chez l'enfant. Les patients atteints de polymyosite peuvent également développer d'autres processus auto-immuns.
Physiopathologie de la dermatomyosite et de la polymyosite
Les modifications pathologiques incluent des lésions cellulaires et une atrophie sur fond d'inflammation plus ou moins grave. Les muscles des membres supérieurs et inférieurs, ainsi que ceux du visage, sont moins touchés que les autres muscles squelettiques. Des lésions des muscles viscéraux du pharynx et de la partie supérieure de l'œsophage, plus rarement du cœur, de l'estomac ou des intestins, peuvent entraîner un dysfonctionnement de ces organes. Des concentrations élevées de myoglobine dues à une rhabdomyolyse peuvent provoquer des lésions rénales. Des modifications inflammatoires des articulations et des poumons peuvent également survenir, en particulier chez les patients porteurs d'anticorps antisynthétases.
Symptômes de la dermatomyosite et de la polymyosite
L'apparition de la polymyosite peut être aiguë (surtout chez l'enfant) ou subaiguë (plus souvent chez l'adulte). Une infection virale aiguë précède parfois la maladie ou en est le déclencheur, les manifestations les plus fréquentes étant une faiblesse des muscles proximaux ou des éruptions cutanées. La douleur est moins prononcée que la faiblesse. Une polyarthralgie, un phénomène de Raynaud, une dysphagie, des troubles pulmonaires et des symptômes généraux (augmentation de la température corporelle, perte de poids, faiblesse) peuvent se développer. Le phénomène de Raynaud est souvent observé chez les patients atteints de maladies concomitantes du tissu conjonctif.
La faiblesse musculaire peut progresser sur plusieurs semaines ou mois. Cependant, pour qu'une faiblesse musculaire se manifeste cliniquement, au moins 50 % des fibres musculaires doivent être atteintes (la présence d'une faiblesse musculaire indique donc la progression d'une myosite). Les patients peuvent éprouver des difficultés à lever les bras au-dessus du niveau des épaules, à monter les escaliers ou à se lever d'une position assise. En raison d'une faiblesse importante des muscles du bassin et de la ceinture scapulaire, les patients peuvent être confinés à un fauteuil roulant ou au lit. Si les muscles fléchisseurs du cou sont touchés, il devient impossible de lever la tête de l'oreiller. L'atteinte des muscles du pharynx et de la partie supérieure de l'œsophage entraîne des troubles de la déglutition et des régurgitations. Les muscles des membres inférieurs, supérieurs et du visage ne sont généralement pas touchés. Cependant, des contractures des membres peuvent se développer.
Les éruptions cutanées observées dans la dermatomyosite sont généralement foncées et érythémateuses. Un œdème périorbitaire violet (éruption héliotrope) est également caractéristique. Les éruptions cutanées peuvent être légèrement surélevées et lisses ou squameuses; elles siègent sur le front, le cou, les épaules, le thorax, le dos, les avant-bras, la partie inférieure des tibias, les sourcils, les genoux, les malléoles internes, les faces dorsales des articulations interphalangiennes et métacarpophalangiennes, et sur la face latérale (symptôme de Gottron). Une hyperhémie de la base ou de la périphérie des ongles est possible. Une dermatite desquamative, accompagnée de crevasses, peut se développer sur la face latérale des doigts. Les lésions cutanées primaires disparaissent souvent sans séquelles, mais peuvent entraîner des modifications secondaires telles qu'une pigmentation foncée, une atrophie, des cicatrices ou un vitiligo. Des calcifications sous-cutanées peuvent se développer, en particulier chez l'enfant.
Environ 30 % des patients développent une polyarthralgie ou une polyarthrite, souvent accompagnée d'œdème et d'épanchement articulaire. Cependant, la gravité des manifestations articulaires est faible. Elles surviennent plus fréquemment lorsque des anticorps anti-Jo-1 ou d'autres synthétases sont détectés.
L'atteinte des organes internes (à l'exception du pharynx et de la partie supérieure de l'œsophage) est moins fréquente dans la polymyosite que dans d'autres maladies rhumatismales (notamment le LES et la sclérodermie systémique). Dans de rares cas, notamment dans le syndrome des antisynthétases, la maladie se manifeste par une pneumopathie interstitielle (se manifestant par une dyspnée et une toux). Des arythmies cardiaques et des troubles de la conduction peuvent se développer, mais ils sont généralement asymptomatiques. Les manifestations gastro-intestinales sont plus fréquentes chez les enfants atteints de vascularite et peuvent inclure des vomissements sanglants, un méléna et une perforation intestinale.
Où est-ce que ça fait mal?
Classification de la polymyosite
Il existe 5 types de polymyosite.
- La polymyosite idiopathique primaire, qui peut survenir à tout âge, n’implique pas la peau.
- La dermatomyosite idiopathique primaire est similaire à la polymyosite idiopathique primaire mais touche la peau.
- La polymyosite et la dermatomyosite associées à des tumeurs malignes peuvent survenir à tout âge; leur apparition est le plus souvent observée chez les patients âgés, ainsi que chez les patients atteints d'autres maladies du tissu conjonctif. Le développement de tumeurs malignes peut être observé dans les deux ans précédant et suivant l'apparition de la myosite.
- La polymyosite ou dermatomyosite infantile est associée à une vascularite systémique.
- La polymyosite et la dermatomyosite peuvent également survenir chez les patients atteints d’autres maladies du tissu conjonctif, le plus souvent la sclérodermie systémique progressive, la maladie du tissu conjonctif mixte et le LES.
L'inclusion de la myosite des muscles troncaux dans le groupe des polymyosites est erronée, car cette dernière est une maladie distincte caractérisée par des manifestations cliniques similaires à celles de la polymyosite chronique idiopathique. Cependant, elle se développe avec l'âge, affecte souvent les muscles des parties distales du corps (par exemple, les membres supérieurs et inférieurs), dure plus longtemps, répond moins bien au traitement et se caractérise par un tableau histologique typique.
Diagnostic de la dermatomyosite et de la polymyosite
Une polymyosite doit être suspectée chez les patients se plaignant d'une faiblesse musculaire proximale, avec ou sans sensibilité. Une recherche de dermatomyosite est nécessaire chez les patients se plaignant d'une éruption cutanée ressemblant à un héliotrope ou au signe de Gottron, ainsi que chez les patients présentant des manifestations de polymyosite associées à des lésions cutanées compatibles avec une dermatomyosite. Les manifestations cliniques de la polymyosite et de la dermatomyosite peuvent ressembler à celles de la sclérodermie systémique ou, plus rarement, du LES ou d'une vascularite. La certitude du diagnostic est renforcée par la réunion d'un maximum des cinq critères suivants:
- faiblesse des muscles proximaux;
- éruptions cutanées caractéristiques;
- augmentation de l'activité des enzymes du tissu musculaire (créatine kinase ou, en l'absence d'augmentation de son activité, aminotransférases ou aldolase);
- modifications caractéristiques à la myographie ou à l'IRM;
- modifications histologiques caractéristiques dans une biopsie de tissu musculaire (critère absolu).
La biopsie musculaire permet d'exclure certaines affections cliniquement similaires, telles que la myosite des muscles troncaux et la rhabdomyolyse due à une infection virale. Les modifications révélées par l'examen histologique peuvent varier, mais une inflammation chronique, des foyers de dégénérescence et de régénération musculaires sont typiques. Un diagnostic précis (généralement par vérification histologique) est nécessaire avant d'initier un traitement potentiellement toxique. L'IRM permet de détecter les foyers d'œdème et d'inflammation musculaires, puis de procéder à une biopsie ciblée.
Les examens de laboratoire peuvent confirmer ou, au contraire, infirmer la suspicion de la maladie, et sont également utiles pour évaluer sa gravité, la possibilité d'une association avec d'autres pathologies similaires et le diagnostic des complications. Bien que des anticorps antinucléaires soient détectés chez certains patients, ce phénomène est plus fréquent dans d'autres maladies du tissu conjonctif. Environ 60 % des patients présentent des anticorps dirigés contre l'antigène des noyaux (PM-1) ou des cellules entières du thymus et contre Jo-1. Le rôle des auto-anticorps dans la pathogenèse de la maladie reste incertain, bien que l'on sache que les anticorps dirigés contre Jo-1 sont un marqueur spécifique du syndrome des antisynthétases, notamment l'alvéolite fibrosante, la fibrose pulmonaire, l'arthrite et le phénomène de Raynaud.
Une évaluation périodique de l'activité de la créatine kinase est utile pour le suivi du traitement. Cependant, chez les patients présentant une atrophie musculaire sévère, l'activité enzymatique peut être normale malgré la présence d'une myosite chronique active. L'IRM, la biopsie musculaire ou une activité élevée de la créatine kinase sont souvent utiles pour différencier une rechute de polymyosite d'une myopathie induite par les glucocorticoïdes.
Étant donné que de nombreux patients présentent des tumeurs malignes non diagnostiquées, certains auteurs recommandent un dépistage chez tous les adultes atteints de dermatomyosite et ceux atteints de polymyosite de plus de 60 ans, selon le protocole suivant: examen physique, incluant examens des seins, du bassin et du rectum (y compris la recherche de sang occulte dans les selles); numération formule sanguine; biochimie sanguine; mammographie; test de l’antigène carcinoembryonnaire; analyse d’urine; radiographie pulmonaire. Certains auteurs s’interrogent sur la nécessité d’un tel dépistage chez les patients plus jeunes, sans signes cliniques de malignité.
Qu'est-ce qu'il faut examiner?
Comment examiner?
Traitement de la dermatomyosite et de la polymyosite
Jusqu'à ce que l'inflammation soit soulagée, l'activité physique doit être limitée. Les glucocorticoïdes sont les médicaments de première intention. Au stade aigu de la maladie, les patients adultes doivent se voir prescrire de la prednisolone (par voie orale) à une dose de 40 à 60 mg par jour. Le dosage régulier de l'activité de la créatine kinase est un indicateur précoce d'efficacité: chez la plupart des patients, une diminution ou une normalisation est observée dans les 6 à 12 semaines suivant une augmentation de la force musculaire. Après normalisation de l'activité enzymatique, la dose de prednisolone est réduite: d'abord d'environ 2,5 mg par jour sur une semaine, puis plus rapidement; en cas de récidive de l'activité enzymatique musculaire, la dose hormonale est à nouveau augmentée. Les patients guéris peuvent se passer de glucocorticoïdes, mais le plus souvent, les adultes nécessitent une corticothérapie au long cours (10 à 15 mg de prednisolone par jour). La dose initiale de prednisolone chez l'enfant est de 30 à 60 mg/m² une fois par jour. En présence d’une rémission > 1 an chez l’enfant, le traitement par glucocorticoïdes peut être interrompu.
Dans certains cas, les patients recevant des doses élevées de glucocorticoïdes ressentent une augmentation soudaine de la faiblesse musculaire, qui peut être associée au développement d’une myopathie glucocorticoïde.
En cas de réponse insuffisante au traitement par glucocorticoïdes, ainsi qu'en cas de développement d'une myopathie glucocorticoïde ou d'autres complications nécessitant une réduction de la dose ou l'arrêt de la prednisolone, des immunosuppresseurs (méthotrexate, cyclophosphamide, azathioprine, ciclosporine) doivent être utilisés. Certains patients peuvent recevoir uniquement du méthotrexate (généralement à des doses supérieures à celles utilisées pour le traitement de la PR) pendant plus de 5 ans. Les immunoglobulines intraveineuses peuvent être efficaces chez les patients réfractaires aux traitements médicamenteux, mais leur utilisation augmente le coût du traitement.
Les myosites associées à des tumeurs primitives et métastatiques, ainsi que les myosites des muscles du tronc, sont généralement plus résistantes à la corticothérapie. La rémission des myosites associées à des tumeurs malignes est possible après ablation de la tumeur.
Quel est le pronostic de la dermatomyosite et de la polymyosite?
Une rémission à long terme (voire une guérison clinique) sur 5 ans est observée chez plus de la moitié des patients traités; ce chiffre est plus élevé chez les enfants. Cependant, une rechute peut survenir à tout moment. Le taux de survie global à cinq ans est de 75 %, plus élevé chez les enfants. Les causes de décès chez l'adulte sont une faiblesse musculaire sévère et progressive, une dysphagie, une malnutrition, une pneumonie par aspiration ou une insuffisance respiratoire due à des infections pulmonaires. La polymyosite est plus grave et résistante au traitement en cas de lésions cardiaques et pulmonaires. Le décès chez l'enfant peut survenir par vascularite intestinale. Le pronostic général de la maladie est également déterminé par la présence de tumeurs malignes.