Expert médical de l'article

Nouvelles publications
Le syndrome d'Angelman chez l'enfant et l'adulte
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.

Il existe un certain nombre de maladies pour lesquelles des expressions comme « prenez soin de vous et vous ne tomberez pas malade » semblent pour le moins ridicules. Il s'agit de pathologies caractérisées par des anomalies mentales et physiques inhérentes au corps de l'enfant avant même sa naissance, mais dont les parents ne sont pas responsables. Ces maladies sont causées par des mutations ou des anomalies chromosomiques et sont dites chromosomiques ou génétiques. Syndrome d'Angelman, syndrome de Down, syndrome de Patau, syndrome d'Edwards, syndrome de Turner, syndrome de Prader-Willi: ce ne sont là que quelques exemples de maladies génétiques parmi une liste assez longue.
Syndrome de l'homme heureux
Cette fois, nous aborderons la pathologie nommée d'après le pédiatre anglais Harry Angelman, qui souleva le premier ce problème en 1965, après avoir rencontré la veille dans son cabinet trois enfants inhabituels, unis par des symptômes communs et particuliers. Le médecin qualifia ces enfants d'« enfants-poupées » et écrivit un article à leur sujet, initialement intitulé « Enfants-marionnettes ». L'article et son titre s'inspirèrent d'un tableau exposé dans un musée de Vérone. Ce tableau représentait un garçon rieur, intitulé « Le Garçon Marionnette ». L'association de l'enfant représenté dans le tableau avec les trois enfants qu'Angelman avait rencontrés dans son cabinet incita le pédiatre à regrouper ces enfants en un seul groupe en raison de leur maladie.
Il n'est pas surprenant que les enfants mentionnés dans l'article n'aient pas été remarqués par d'autres médecins. Après tout, à première vue, il semblait qu'ils souffraient de maladies complètement différentes, tant le tableau clinique général était différent dans trois cas différents. Cette « nouvelle » pathologie chromosomique aurait peut-être intéressé d'autres scientifiques, mais à cette époque, la génétique n'était pas encore suffisamment développée pour confirmer l'hypothèse du médecin anglais. C'est pourquoi, après un certain intérêt, l'article a été longtemps mis de côté.
La mention suivante du syndrome d'Angelman, nom donné à l'article du pédiatre anglais G. Angelman, remonte au début des années 1980. Ce n'est qu'en 1987 que l'on a pu comprendre pourquoi une petite proportion d'enfants naissent avec des anomalies telles qu'ils semblent constamment sourire et heureux. En réalité, c'est totalement faux: le sourire n'est qu'une grimace derrière laquelle se cachent le malheur et la douleur des parents.
Épidémiologie
Selon les statistiques, une mutation chromosomique peut se développer chez un enfant, que ses parents présentent des mutations similaires ou non. Il n'existe pas de caractère héréditaire clair du syndrome d'Angelman (SA), mais la probabilité de développer une pathologie chez des parents porteurs de mutations chromosomiques est assez élevée.
Il est également intéressant de noter que si une famille a déjà un enfant atteint du SA, il y a un pour cent de risque d’avoir un deuxième enfant atteint du même trouble, même si les parents sont en bonne santé.
Il n'existe pas encore de statistiques précises sur le nombre de patients atteints du syndrome d'Angelman. La raison pourrait être la diversité des symptômes, qui peuvent apparaître de manière spécifique ou ne pas apparaître du tout pendant une longue période. On estime que la prévalence de la maladie est d'un enfant pour 20 000 nouveau-nés. Ce chiffre est toutefois très approximatif.
Causes Syndrome d'Angelman
Le syndrome d'Angelman est le nom médical d'une pathologie chromosomique, mais il est loin d'être le seul. On appelle cette maladie le syndrome des enfants-poupées, le syndrome de la marionnette heureuse, le syndrome de Petrouchka ou encore le syndrome de la poupée rieuse. On invente toutes sortes de noms (parfois offensants pour les patients eux-mêmes et leurs parents), mais une maladie est une maladie, aussi drôle soit-elle et quelles qu'en soient les causes.
Le syndrome d'Angelman, comme de nombreuses autres pathologies génétiques, est toujours dû à des anomalies structurelles d'un chromosome ou de l'ensemble des chromosomes. Dans notre cas, le problème réside dans le chromosome 15, transmis par la mère. Autrement dit, le chromosome paternel ne présente aucune anomalie, tandis que le chromosome féminin subit certaines mutations.
Selon le type d'anomalie chromosomique, le syndrome d'Angelman est classé comme une mutation chromosomique. Ces mutations sont considérées comme:
- Une délétion (absence d'une section d'un chromosome contenant un certain ensemble de gènes; si l'un des gènes est manquant, on parle de microdélétion), qui est le résultat de deux cassures et d'une réunion, lorsqu'une section du chromosome d'origine est perdue.
- Duplication (présence d'une section supplémentaire dans un chromosome qui est une copie d'un chromosome existant), qui dans la plupart des cas conduit à la mort d'une personne, et moins souvent à l'infertilité.
- Inversion (inversion de l'une des sections du chromosome de 180 degrés, c'est-à-dire dans la direction opposée, et les gènes qu'il contient sont alors situés dans l'ordre opposé), lorsque les extrémités cassées du chromosome sont connectées dans un ordre différent de l'original.
- Insertion (si une partie du matériel génétique d'un chromosome est déplacée),
- translocation (si une certaine section d'un chromosome est attachée à un autre chromosome; une telle mutation peut être mutuelle sans perte de sections).
Recevant un chromosome muté d'une mère sans méfiance, l'enfant est condamné à naître avec des anomalies. La cause la plus fréquente du syndrome d'Angelman est encore considérée comme une délétion du 15e chromosome maternel, lorsqu'une petite section est manquante. Les mutations moins fréquentes du syndrome de la « poupée rieuse » sont:
- translocation,
- disomie unipaternelle (si l'enfant a reçu une paire de chromosomes du père, le chromosome maternel est absent),
- mutation de gènes dans l'ADN, qui constituent à la fois le principal matériau de construction (génétique) et les instructions pour son utilisation correcte (en particulier, mutation du gène ube3a dans le chromosome maternel).
La présence d'une de ces mutations chez les parents constitue un facteur de risque de développement du syndrome d'Angelman chez l'enfant. Cependant, les mutations chromosomiques ne sont pas les seules à pouvoir provoquer le développement de la maladie chez l'enfant. Les mutations génomiques (associées à une modification quantitative des chromosomes et plus fréquentes) sont également susceptibles de provoquer cette maladie. Parmi les mutations génomiques les plus fréquentes, on trouve la trisomie chromosomique (lorsque le chromosome d'une personne comporte plus de 46 chromosomes).
Pour qu'une pathologie se manifeste chez un enfant, il n'est pas nécessaire que les parents présentent des anomalies chromosomiques. Pourtant, chez un certain pourcentage de patients, la maladie est héréditaire.
Pathogénèse
Plongeons-nous un peu plus profondément dans la biologie, ou plus précisément dans la génétique. L'information génétique de chaque organisme humain est contenue dans 23 paires de chromosomes. Un chromosome d'une paire est transmis à l'enfant par le père, l'autre par la mère. Toutes les paires de chromosomes diffèrent par leur forme et leur taille et contiennent certaines informations. Ainsi, la 23e paire de chromosomes (chromosomes X et Y) est responsable de la formation des caractères sexuels du bébé (XX = fille, XY = garçon, tandis que le chromosome Y ne peut être transmis à l'enfant que par le père).
Idéalement, un enfant reçoit 46 chromosomes de ses parents, qui constituent ses caractéristiques génétiques et le prédéterminent en tant qu'individu. Un nombre supérieur de chromosomes est appelé trisomie et est considéré comme une déviation de la norme. Par exemple, la présence du chromosome 47 dans l'ensemble chromosomique (caryotype, déterminant l'espèce et les caractéristiques individuelles) est à l'origine du syndrome de Down.
Si les chromosomes sont colorés avec un colorant spécial, on peut observer au microscope des bandes de différentes nuances le long de chacun d'eux. À l'intérieur de chaque bande se trouve un grand nombre de gènes. Toutes ces bandes sont numérotées par les scientifiques et ont une position fixe. L'absence d'une de ces bandes est considérée comme une anomalie. Dans le syndrome d'Angelman, on observe très souvent l'absence de segments du chromosome maternel dans l'intervalle q11-q13, situé dans le bras long, dont le nombre de bases d'ADN n'est que d'environ 4 millions.
Le composant principal du chromosome est considéré comme une molécule d'ADN incroyablement longue contenant des milliers de gènes et des dizaines, voire des centaines de millions de bases azotées. Ainsi, le chromosome 15, responsable du développement du syndrome d'Angelman et de plusieurs autres, contient 1 200 gènes et environ 100 millions de bases. Toute altération de la structure de la molécule d'ADN affectera certainement l'apparence et le développement du futur enfant.
L'information génétique contenue dans les gènes est convertie en protéines ou en ARN. Ce processus est appelé expression génétique. De cette façon, l'information génétique reçue des parents acquiert à la fois forme et contenu, qui sont incarnés par leur héritier unique, féminin ou masculin.
Il existe un certain nombre de pathologies avec un type d'hérédité non classique, notamment le syndrome d'Angelman, dans lequel les gènes reçus des parents sous forme de chromosomes appariés portent une empreinte unique des parents et se manifestent de différentes manières.
Le syndrome d'Angelman est un exemple frappant d'empreinte génomique, où l'expression des gènes dans l'organisme de l'enfant dépend directement du parent dont les allèles ont été reçus (formes différentes d'un même gène, reçues du père et de la mère, situées sur des sections identiques de chromosomes appariés). Autrement dit, seules les anomalies du chromosome maternel conduisent au développement du syndrome, tandis que les mutations et les anomalies structurelles du chromosome paternel provoquent des pathologies complètement différentes.
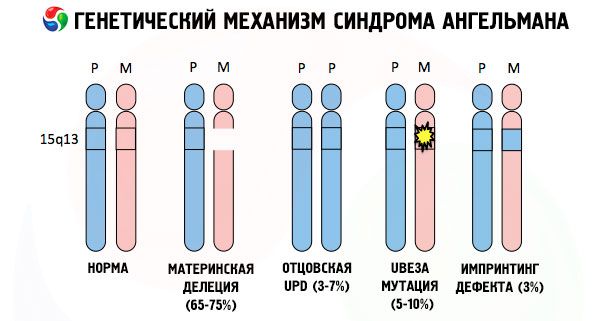
Dans cette pathologie, on observe une absence de certains gènes du chromosome maternel ou une perte/réduction de l'activité de certains gènes (dans la grande majorité des cas, le gène ube3a, impliqué dans le métabolisme de l'ubiquitine, une protéine régulant la dégradation d'autres protéines). Par conséquent, l'enfant est diagnostiqué avec des anomalies du développement mental et des malformations physiques.
Symptômes Syndrome d'Angelman
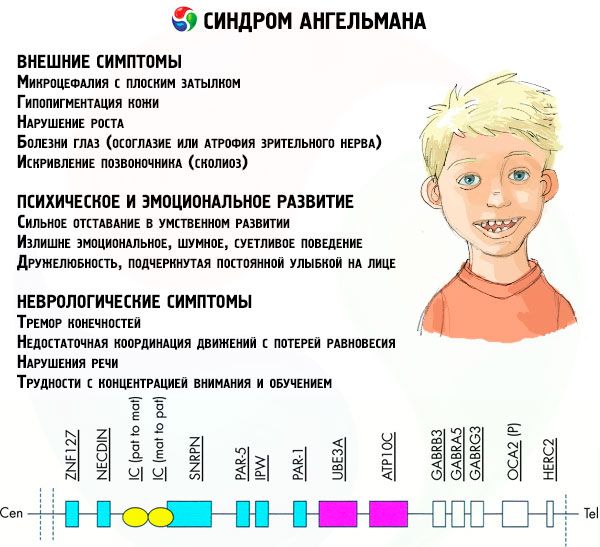
Les symptômes du syndrome d'Angelman affectent divers aspects de la vie et du développement de l'enfant: physiques, neurologiques et mentaux. Sur cette base, trois groupes de symptômes peuvent être identifiés, indiquant le développement de cette pathologie.
- Symptômes externes ou physiques:
- une tête disproportionnée par rapport au corps et aux membres, qui sont de taille normale,
- bouche trop large,
- il y a presque toujours un sourire sur le visage (avec la bouche ouverte),
- dents clairsemées,
- lèvre supérieure étroite,
- langue souvent saillante et large,
- mâchoire inférieure saillante,
- menton pointu,
- peau très claire, souvent poilue (albinisme, lié au fait que le corps ne produit pas le pigment mélanine),
- taches brunes sur peau claire (hypopigmentation due à une production insuffisante de mélanine)
- symptômes physiques ou externes: maladies oculaires telles que le strabisme ou l'atrophie du nerf optique,
- courbure de la colonne vertébrale (scoliose),
- jambes raides (lors de la marche, une personne ne plie pas ses jambes au niveau des genoux en raison d'une faible mobilité des articulations, d'où la comparaison avec la démarche d'une poupée).
- Symptômes liés au développement mental et émotionnel:
- retard mental sévère,
- comportement trop émotif, bruyant et difficile,
- applaudissements fréquents des mains,
- exprimé une gentillesse, soulignée par un sourire constant sur le visage,
- rires fréquents sans raison.
- Symptômes neurologiques:
- tremblement des membres,
- coordination insuffisante des mouvements avec perte d'équilibre,
- diminution du tonus musculaire,
- divers troubles du sommeil,
- crises hystériques fréquentes dans l'enfance,
- troubles de la parole (l'enfant commence à parler tard, a de mauvaises capacités de communication et a des troubles de l'élocution),
- hyperactivité sur fond d'excitabilité accrue,
- difficultés de concentration et d'apprentissage.
Il s'agit toutefois d'une description générale de la maladie. En réalité, le tableau clinique du syndrome d'Angelman dépend largement du stade d'évolution de la maladie et du type de mutation chromosomique à l'origine de la pathologie. Cela signifie que les symptômes peuvent varier considérablement d'un patient à l'autre, ce qui a longtemps empêché de distinguer cette pathologie d'autres présentant un tableau clinique similaire.
Parmi le nombre total de symptômes, nous pouvons souligner ceux qui sont caractéristiques de tous les patients sans exception:
- retard mental sévère,
- comportement inapproprié (rire déraisonnable, excitabilité accrue, manque de concentration, état d'euphorie),
- sous-développement des capacités motrices,
- mauvaise coordination des mouvements, ataxie de la marche (allure irrégulière, balancement latéral, etc.), tremblement des membres.
- trouble du développement de la parole avec prédominance des moyens de communication non verbaux.
Parmi les symptômes rencontrés par la grande majorité des patients, on peut distinguer:
- disproportion entre la tête et le corps causée par un retard de développement physique,
- chez de nombreux patients, la forme du crâne est telle que la taille du cerveau reste plus petite que chez les personnes en bonne santé (microcéphalie),
- crises d'épilepsie avant l'âge de 3 ans avec une diminution progressive de la force et de la fréquence à un âge plus avancé,
- distorsion des paramètres EEG (fluctuations et amplitude élevée des ondes de basse fréquence).
Ces symptômes sont assez courants, cependant, 20 % des patients atteints du syndrome d’Angelman n’en présentent pas.
Encore moins fréquemment, il est possible de diagnostiquer des manifestations de la maladie telles que:
- strabisme sévère ou léger,
- un mauvais contrôle du mouvement de la langue, ce qui fait que les patients tirent souvent la langue sans raison,
- difficultés de déglutition et de succion, en particulier chez les jeunes enfants,
- perturbation de la pigmentation de la peau et des yeux,
- bras levés ou pliés en marchant,
- hyperréflexie,
- troubles du sommeil, en particulier chez l'enfant,
- salivation fréquente,
- soif insatiable,
- mouvements de mastication trop actifs,
- hypersensibilité à la chaleur,
- arrière de la tête plat,
- mâchoire inférieure saillante,
- paumes lisses.
Un pourcentage important de patients présentent des problèmes de miction, qu'ils contrôlent mal, une altération de la motricité fine, ce qui entraîne des difficultés d'autonomie et d'apprentissage, ainsi qu'un excès de poids. Presque tous les patients ont une puberté plus tardive que leurs pairs en bonne santé.
Les enfants atteints du syndrome d'Angelman perçoivent bien la parole et la comprennent, mais ne souhaitent pas participer aux conversations, limitant leur discours à quelques dizaines de mots nécessaires à la vie quotidienne. Cependant, à l'âge adulte, ces patients paraissent plus jeunes que leurs pairs sans pathologie génétique.
De nombreux symptômes du syndrome d'Angelman sont instables; le tableau clinique de la maladie évolue donc considérablement avec l'âge. Les convulsions et les crises d'épilepsie deviennent moins fréquentes, voire disparaissent complètement, le patient devient moins excitable et son sommeil s'améliore.
Complications et conséquences
Le syndrome d'Angelman est une pathologie chromosomique grave, actuellement pratiquement incurable, qui prive les patients de la possibilité de vivre une vie normale. La vie d'un enfant atteint du SA dépend en grande partie du type d'anomalie chromosomique.
Dans la plupart des cas, la duplication d'un segment chromosomique est incompatible avec la vie. Et même si ces patients ne meurent pas en bas âge et atteignent la puberté, ils n'ont aucune chance d'avoir des enfants.
La délétion ou l'absence d'une partie des gènes, fréquente dans le syndrome d'Angelman, constitue un obstacle à l'apprentissage de la marche et de la parole. Ces enfants présentent un retard mental plus sévère, et les crises d'épilepsie sont plus fréquentes et d'intensité bien supérieure à celles des patients présentant d'autres anomalies chromosomiques.
S'il n'y a qu'une mutation d'un seul gène, avec une attention et une approche appropriées, l'enfant peut apprendre les bases de l'auto-soin, de la communication et de l'interaction dans un groupe, même s'il sera toujours en retard par rapport à ses pairs en termes de développement.
Pour les enfants atteints du syndrome d'Angelman, naturellement bienveillants, l'amour et l'attention de leurs parents sont essentiels. C'est seulement dans ce cas que leur éducation portera ses fruits, même modestement. Bien entendu, les patients atteints du syndrome d'Asperger ne pourront pas étudier dans une école ordinaire. Ils ont besoin de classes spéciales où l'on apprendra d'abord à se concentrer, puis où l'on leur transmettra progressivement les bases du savoir scolaire.
Diagnostics Syndrome d'Angelman
Le syndrome d'Angelman est une pathologie congénitale du développement. Cependant, en raison de certaines circonstances, son diagnostic est souvent impossible chez le nourrisson et la petite enfance. Cela est dû à la non-spécificité et à la faible expression des symptômes chez les nourrissons et les enfants de moins de 3 ans. De plus, la prévalence de la maladie dans notre pays n'est pas suffisamment élevée pour que les médecins puissent la reconnaître parmi ses pairs.
Le syndrome d'Angelman chez le nourrisson peut se manifester par une diminution du tonus musculaire, qui se traduit par des difficultés d'alimentation (faiblesse du réflexe de succion et de déglutition), puis par des difficultés d'apprentissage de la marche (ces enfants commencent à marcher beaucoup plus tard). Ces symptômes sont les premiers signes d'une anomalie du développement du bébé, qui pourrait être associée à une anomalie chromosomique. Seule une analyse génétique peut confirmer cette hypothèse.
Une attention particulière est portée aux enfants dont les parents présentent diverses anomalies génomiques ou chromosomiques. En effet, la maladie peut ne pas se manifester immédiatement, et si elle est détectée à temps, un travail intensif avec l'enfant peut améliorer considérablement ses résultats scolaires et ralentir sa progression.
Si les parents présentent diverses anomalies chromosomiques, une analyse génétique est effectuée avant même la naissance du bébé, car la SA est l'une des pathologies qui peuvent être détectées au stade embryonnaire.
La collecte de matériel pour la recherche génétique peut être réalisée de deux manières:
- invasive (avec un certain pourcentage de risque, car il est nécessaire de pénétrer l'utérus pour prélever un échantillon de liquide amniotique),
- non invasif (analyse de l'ADN du bébé à partir du sang de la mère).
Les études suivantes sont ensuite réalisées:
- hybridation in situ fluorescente (méthode FISH) – liaison d’une sonde d’ADN marquée avec un colorant spécial à l’ADN étudié, suivie d’un examen au microscope.
- analyse des mutations du gène ube3a et des gènes imprimés,
- Analyse de la méthylation de l'ADN à l'aide de méthodes spéciales utilisées en génétique.
Les tests génétiques fournissent des informations relativement précises en cas d'anomalies chromosomiques, ce qui permet aux futurs parents de savoir à l'avance à quoi se préparer. Il existe toutefois des exceptions. Chez certains patients, en présence de tous les symptômes évoquant une pathologie, les résultats des tests restent normaux. Autrement dit, la pathologie ne peut être identifiée qu'en observant attentivement l'enfant dès sa plus tendre enfance: comment il mange, quand il a commencé à marcher et à parler, s'il plie les jambes en marchant, etc.
Outre la méthode FISH, parmi les méthodes diagnostiques instrumentales du syndrome d'Angelman, on peut citer la tomographie (TDM ou IRM), qui permet de déterminer l'état et la taille du cerveau, et l'électroencéphalogramme (EEG), qui montre comment fonctionnent les différentes parties du cerveau.
Les médecins posent généralement un diagnostic définitif à l'âge de 3 à 7 ans, lorsque le patient présente déjà la plupart des symptômes et que la dynamique du développement de la maladie est visible.
Quels tests sont nécessaires?
Diagnostic différentiel
Le syndrome d'Angelman est une pathologie génétique pratiquement inexistante. La plupart des symptômes peuvent indiquer à la fois une SA et d'autres pathologies génétiques.
Le diagnostic différentiel du syndrome d'Angelman est réalisé avec les pathologies suivantes:
- Syndrome de Pitt-Hopkins (les patients se caractérisent par un retard mental, un caractère joyeux, souriant, une bouche plutôt grande et large, et une microcéphalie). La différence réside dans des crises d'hyperventilation et des apnées à l'état de veille.
- Syndrome de Christianson (les patients sont des personnes mentalement retardées avec un tempérament joyeux, incapables de parler, caractérisées par une microcéphalie, une ataxie, des convulsions, des mouvements musculaires involontaires).
- Syndrome de Mowat-Wilson (symptômes: retard mental, crises d’épilepsie, menton pointu, bouche ouverte, expression joyeuse du visage, microcéphalie). Particularités: grand écartement des yeux, yeux bridés vers l’intérieur, pointe du nez arrondie, pavillon de l’oreille tourné vers l’arrière.
- Syndrome de Kabuki (caractérisé par un retard mental léger à modéré, des troubles de la parole et de la motricité, une faiblesse musculaire, des crises d'épilepsie, une microcéphalie, de longs intervalles entre les démangeaisons et une altération de la coordination). Il se caractérise par des sourcils arqués, une paupière inférieure latéralement éversée, des yeux écartés, de longues fentes palpébrales et des cils longs et épais.
- Syndrome de Rett (différenciation du syndrome de Rett chez la femme). Symptômes: retard du développement du langage, convulsions, microcéphalie. La différence réside dans l'absence d'expression joyeuse du visage, avec des crises d'apnée et d'apraxie, qui progressent avec le temps.
- Syndrome de retard mental autosomique récessif 38 (symptômes: retard mental marqué avec retards moteurs et de langage, faiblesse musculaire, troubles de l'alimentation pendant la petite enfance, impulsivité). Le signe distinctif est la couleur bleue de l'iris.
- Syndrome de duplication du gène MECP2 (différenciation du SA chez les garçons). Symptômes: retard mental sévère, faiblesse musculaire depuis l'enfance, troubles ou absence de langage, épilepsie. Particularités: myopathie progressive, infections récurrentes.
- Syndrome de Kleefstra (symptômes: troubles de la parole et de la pensée, faiblesse musculaire, troubles du sommeil, manque d’attention, bouche ouverte, hyperactivité, convulsions, ataxie, troubles de l’équilibre). Signes distinctifs: visage plat, nez court et retroussé, yeux écartés, lèvre inférieure éversée et large, accès d’agressivité.
- Syndrome de Smith-Magenis (caractérisé par des convulsions, des troubles du sommeil et des troubles du développement intellectuel et moteur). Les signes distinctifs comprennent un visage large et plat et un front proéminent.
- Syndrome de Koolen-de Vries (retard mental léger à modéré, faiblesse musculaire, convulsions, sociabilité). Signes distinctifs: visage allongé avec front haut, oreilles décollées, yeux bridés, grande mobilité articulaire, malformations cardiaques congénitales.
- Syndrome de Phelan-McDermid (symptômes: retard mental, troubles de la parole ou absence de langage). Particularités: grandes mains avec muscles développés, faiblesse musculaire dès la naissance, faible transpiration.
Des pathologies telles que le déficit en adénylsuccinate, le syndrome de retard mental autosomique récessif 1, le syndrome de duplication du chromosome 2q23.1, les syndromes d'haploinsuffisance des gènes FOXG1, STXBP1 ou MEF2C et quelques autres peuvent « se vanter » de symptômes similaires au syndrome d'Angelman.
La tâche du médecin est d'établir un diagnostic précis, en différenciant le syndrome d'Angelman des pathologies présentant des symptômes similaires, et de prescrire un traitement efficace adapté au stade diagnostiqué de la maladie.
Qui contacter?
Traitement Syndrome d'Angelman
Le syndrome d'Angelman fait partie de ces pathologies pour lesquelles la médecine est encore à la recherche d'un traitement efficace. Le traitement étiologique de la maladie est en cours de développement, avec diverses méthodes et moyens, dont beaucoup n'ont pas encore été testés sur l'homme. Cela signifie que, pour l'instant, les médecins doivent se limiter à un traitement symptomatique, qui contribue à soulager la situation peu enviable des enfants et des adultes atteints du syndrome de la marionnette, souffrant de crises d'épilepsie, de salivation excessive, d'hypotension et de troubles du sommeil.
Il est donc possible de réduire la fréquence et l'intensité des crises d'épilepsie grâce à un anticonvulsivant bien choisi. Cependant, la difficulté réside dans le fait que les crises d'épilepsie chez les patients atteints d'AS diffèrent des crises d'épilepsie classiques en ce qu'elles se caractérisent par plusieurs types de crises, ce qui permet de soulager la maladie en administrant plusieurs médicaments simultanément.
Les anticonvulsivants les plus couramment utilisés pour traiter le syndrome d'Angelman sont l'acide valproïque, le topiramate, la lamotrigine, le lévétiracétam, le clonazépam et les médicaments à base de ces médicaments. Les médicaments à base de carmazépine, de phénytoïne, de phénobarbital et d'éthosuximide sont moins fréquemment utilisés, car certains d'entre eux peuvent provoquer un effet paradoxal consistant à intensifier et à augmenter la fréquence des crises d'épilepsie. Ce phénomène se produit en cas de monothérapie.
Pour traiter la salivation excessive, deux méthodes sont généralement utilisées: médicamenteuses (médicaments qui inhibent la production de salive) et chirurgicales, qui impliquent la réimplantation des canaux salivaires. Cependant, dans le cas de l'AS, ces méthodes sont considérées comme inefficaces et la question reste ouverte. Les parents et les personnes qui s'occupent de ces patients doivent y prêter une attention particulière, car les patients eux-mêmes ne contrôlent généralement pas leur salivation, et certains sont tout simplement incapables de la gérer eux-mêmes.
Un autre problème est la courte durée du sommeil. Souvent, les enfants atteints du syndrome d'Angelman ne dorment pas plus de 5 heures, ce qui a un impact négatif sur le fonctionnement de l'organisme. Les enfants actifs, facilement excitables, qui aiment les jeux et la communication (même s'ils se limitent aux méthodes non verbales) sont visiblement fatigués pendant la journée. Pour bien se reposer, le corps a besoin d'un sommeil profond et complet, mais c'est précisément là que réside le problème.
Il semblerait que les sédatifs (phénothiazines et antipsychotiques atypiques) qui calment le système nerveux devraient suffire à améliorer le sommeil des patients excitables. Cependant, dans le cas du SA, leur utilisation est susceptible d'entraîner des effets indésirables. Par conséquent, les médecins privilégient toujours les somnifères légers, comme la mélatonine (un médicament hormonal naturel à base d'hormone du sommeil), administrée une heure avant le coucher à raison d'un comprimé, et la diphénhydramine. La fréquence d'administration et la posologie sont déterminées par le médecin en fonction de l'état et de l'âge du patient.
Les patients atteints du syndrome d'Angelman présentent parfois des troubles digestifs et des selles difficiles. Des laxatifs (de préférence à base de plantes) peuvent améliorer la qualité de leurs selles.
On peut aussi aborder le problème différemment, comme l'ont fait les médecins américains, en s'appuyant sur certaines méthodes de traitement de l'autisme, car de nombreux symptômes caractéristiques du SA le sont également (impulsivité, mouvements involontaires, actions répétitives, déficit de l'attention, problèmes de communication, etc.). Il a été constaté que l'introduction de l'hormone sécrétine, qui normalise la digestion et les selles, a un effet positif sur l'attention des patients, et que l'ocytocine contribue à améliorer les capacités cognitives et la mémoire de l'enfant, ainsi qu'à corriger son comportement.
Certes, les hormones seules ne suffisent pas, surtout chez les enfants. Dans le syndrome d'Angelman, une thérapie comportementale, un suivi psychologique et une orthophonie (apprentissage des méthodes de communication non verbale et de la langue des signes) sont indiqués. L'éducation de ces enfants doit s'appuyer sur un programme personnalisé avec la participation d'enseignants spécialement formés, d'un psychologue et des parents. Malheureusement, ce n'est pas possible partout et les familles sont laissées seules face à leur problème.
Étant donné que de nombreux jeunes patients atteints de SA souffrent d'un faible tonus musculaire et de problèmes articulaires, la physiothérapie est une priorité. Le plus souvent, les médecins ont recours à des applications de paraffine, à l'électrophorèse et à la magnétothérapie.
Un massage tonique actif et des exercices spécifiques de rééducation physique aideront l'enfant malade à se tenir debout et à marcher avec assurance au bout d'un certain temps. L'aquagym, recommandée pour l'AS en eau fraîche, est particulièrement utile à cet égard. Elle renforce le tonus musculaire et apprend à l'enfant à contrôler son corps et à coordonner ses mouvements.
Traitement anticonvulsivant
Le symptôme le plus dangereux du syndrome d'Angelman est la survenue de crises d'épilepsie. Ce symptôme est observé chez 80 % des patients, ce qui nécessite un traitement anticonvulsivant efficace.
Le traitement des crises d'épilepsie repose sur la prise de vitamines et d'anticonvulsivants. En cas de syndrome d'Angelman, accompagné d'un syndrome convulsif, les vitamines B, C, D et E sont utiles. Cependant, prescrire soi-même une vitaminothérapie dans ce cas est très dangereux, car un apport incontrôlé de vitamines peut réduire l'efficacité des antiépileptiques et provoquer de nouvelles crises plus graves et prolongées.
Le choix des anticonvulsivants et la prescription de leur dosage efficace doivent également être effectués par un médecin spécialiste. Il ou elle déterminera également si un seul médicament sera suffisant ou si le patient devra en prendre deux ou plusieurs pendant une longue période.
Pour la plupart des patients, les médecins prescrivent des médicaments à base d’acide valproïque (acide valproïque, Dépakine, Convulex, Valparine, etc.), qui préviennent les crises et améliorent l’humeur et l’état mental des patients.
L'acide valproïque est disponible sous forme de comprimés, de sirop et de solutions injectables. Le médicament le plus populaire est la « Dépakine » à libération prolongée, en comprimés et en solution pour administration intraveineuse. La posologie est déterminée individuellement par le médecin en fonction du poids, de l'âge et de l'état de santé du patient.
Le médicament est pris au cours des repas, 2 à 3 fois par jour. La dose quotidienne moyenne est de 20 à 30 mg par kilogramme de poids corporel, la dose maximale étant de 50 mg/kg par jour.
Contre-indications d'utilisation. Ne pas utiliser en cas de dysfonctionnement hépatique et pancréatique, de diathèse hémorragique, d'hépatite, de porphyrie et d'hypersensibilité au médicament.
Les effets secondaires comprennent des tremblements des mains, des troubles digestifs et des selles, ainsi que des changements de poids corporel.
Le « topiramate » est également un médicament de choix pour l'AS. Il est présenté sous forme de comprimés et utilisé en monothérapie ou en association avec d'autres médicaments.
Mode d'administration et posologie. Prendre les comprimés par voie orale, indépendamment des repas. La dose quotidienne initiale est de 25 à 50 mg pour les adultes et de 0,5 à 1 mg/kg pour les enfants. La dose est augmentée chaque semaine selon les recommandations du médecin.
Ce médicament ne doit pas être pris pendant la grossesse et l'allaitement, ni en cas d'hypersensibilité à ses composants. Il présente de nombreux effets secondaires.
Médicaments qu'un médecin peut prescrire pour le syndrome d'Angelman: Clomazépam, Rivotril, Lamotrigine, Seizar, Lamictal, Lévétiracétam, Keppra, Epiterra, etc.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Médecine traditionnelle et homéopathie
La médecine traditionnelle, comme les préparations homéopathiques, sont bien sûr relativement sûres, mais l’efficacité d’un tel traitement pour le syndrome d’Angelman peut être considérée comme controversée.
Bien que les traitements traditionnels puissent encore aider dans certains domaines, notamment pour stopper les crises d'épilepsie, les traitements à base de plantes peuvent s'avérer très efficaces.
Un mélange médicinal à base de pivoine, de réglisse et de lentille d'eau (à prendre en quantités égales) procure un effet bénéfique. Les herbes doivent être moulues en farine. Deux semaines après le début de la prise, on constate une diminution significative de la fréquence des crises.
La décoction de lavande (1 cuillère à café par verre d'eau bouillante) est également efficace contre les crampes. Le mélange est bouilli pendant 5 minutes et infusé pendant une demi-heure. Le médicament est pris le soir pendant 14 jours.
Une infusion aqueuse (ou alcoolisée) d'agripaume est considérée comme efficace contre les crises d'épilepsie.
Parmi les préparations homéopathiques pour prévenir les crises d'épilepsie dans le syndrome d'Angelman, on peut utiliser des médicaments à base de camomille et d'agripaume, d'Acidum hydrocyanicum, d'Argentum nitricum, de Kalium bromatum et d'Arsenicum album. Il est toutefois important de noter que seul un homéopathe peut prescrire des doses efficaces et sûres de médicaments dans chaque cas spécifique.
La prévention
Comme le lecteur l'a probablement déjà compris, la médecine n'est pas encore en mesure de prévenir les mutations génétiques et autres anomalies chromosomiques, ni de corriger la situation. Cela peut arriver à n'importe qui, car les enfants atteints du syndrome d'Angelman naissent de parents en bonne santé, et la génétique, actuellement l'une des branches les moins étudiées de la médecine, ne peut pas encore l'expliquer.
La seule chose à faire est d'adopter une approche responsable en matière de planification de grossesse, de s'inscrire et de se soumettre aux examens à temps. Mais, encore une fois, une telle mesure sera plus éducative que préventive, comme tout examen. Les jeunes parents sauront à l'avance à quoi se préparer et, en cas de réponse positive, ils décideront s'ils peuvent assumer la responsabilité d'élever un enfant malade.
Prévoir
Le pronostic du syndrome d'Angelman dépend de la nature de l'anomalie chromosomique et de la rapidité de son dépistage. Les enfants les plus touchés sont ceux dont le chromosome 15 présente des « trous » génétiques (délétion). La probabilité que ces patients puissent marcher et parler est extrêmement faible. D'autres cas peuvent être corrigés grâce à une approche attentive et à l'amour de votre enfant.
Malheureusement, ces patients ne pourront pas s'intégrer pleinement à la société, même s'ils sont loin d'être stupides et comprennent la parole et son sens. Cependant, ils auront des difficultés de communication toute leur vie. On peut leur apprendre la langue des signes dès l'enfance, mais on ne peut pas les forcer à communiquer avec des mots. Le vocabulaire des patients « parlants » se limite au minimum de mots utilisés dans la vie quotidienne (5 à 15 mots).
Quant à l'espérance de vie et à l'état de santé général des patients atteints du syndrome d'Angelman, les chiffres oscillent autour des valeurs moyennes. À l'âge adulte, les patients sont principalement confrontés à des problèmes de santé tels que la scoliose et l'obésité, qui, avec un traitement adapté, ne mettent pas leur pronostic vital en danger.