Expert médical de l'article

Nouvelles publications
Syndrome de Treacher Collins
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.

Les troubles intra-utérins des processus de développement osseux provoquent de graves déformations cranio-faciales, et l'une des variétés de cette pathologie est le syndrome de Treacher Collins (TCS) ou dysostose mandibulofasciale, c'est-à-dire maxillo-faciale.
Code de la maladie selon la CIM 10: classe XVII (anomalies congénitales, déformations et troubles chromosomiques), Q75.4 - dysostose mandibulo-faciale.
Causes Syndrome de Treacher Collins
Ce syndrome doit son nom à l'éminent ophtalmologiste britannique Edward Treacher Collins, qui en a décrit les principales caractéristiques il y a plus de cent ans. Cependant, les médecins européens appellent plus souvent ce type d'anomalie des os de la face et de la mâchoire « maladie ou syndrome de Franceschetti », en s'appuyant sur les recherches approfondies de l'ophtalmologiste suisse Adolf Franceschetti, qui a introduit le terme « dysostose mandibulofasciale » au milieu du siècle dernier. Dans le milieu médical, on parle également de syndrome de Franceschetti-Collins.
Le syndrome de Treacher Collins est causé par des mutations du gène TCOF1 (locus chromosomique 5q31.3-33.3), qui code pour une phosphoprotéine nucléolaire responsable de la formation de la partie craniofaciale de l'embryon humain. Suite à une diminution prématurée de la quantité de cette protéine, la biogenèse et les fonctions de l'ARNr sont perturbées. Selon les généticiens du programme de recherche sur le génome humain, ces processus entraînent une réduction de la prolifération des cellules embryonnaires de la crête neurale – une crête le long du sillon neural qui se ferme pour former le tube neural au cours du développement embryonnaire.
La formation des tissus faciaux résulte de la transformation et de la différenciation des cellules de la partie supérieure (tête) de la crête neurale, qui migrent le long du tube neural vers la zone des premier et deuxième arcs branchiaux de l'embryon. La déficience de ces cellules entraîne des déformations craniofaciales. La période critique d'apparition des anomalies se situe entre 18 et 28 jours après la fécondation. Une fois la migration des cellules de la crête neurale terminée (quatrième semaine de gestation), la quasi-totalité des tissus mésenchymateux lâches de la région faciale se forment. Ces tissus se différencient ensuite (entre 5 et 8 semaines) en tissus squelettiques et conjonctifs de toutes les parties du visage, du cou, du larynx, de l'oreille (y compris l'oreille interne) et des futures dents.
Pathogénèse
La pathogénèse du syndrome de Treacher Collins est souvent familiale et l'anomalie est transmise selon le mode autosomique dominant, bien qu'il existe des cas de transmission autosomique récessive (avec des mutations d'autres gènes, notamment POLR1C et POLR1D). Le plus imprévisible concernant la dysostose maxillo-faciale est que la mutation n'est transmise aux enfants que dans 40 à 48 % des cas. Autrement dit, chez 52 à 60 % des patients, les causes du syndrome de Treacher Collins ne sont pas liées à la présence d'une anomalie dans la famille, et on pense que la pathologie résulte de mutations génétiques sporadiques de novo. Les nouvelles mutations sont très probablement la conséquence d'effets tératogènes sur le fœtus pendant la grossesse.
Parmi les causes tératogènes de ce syndrome, les experts citent de fortes doses d'éthanol (alcool éthylique), les radiations, la fumée de cigarette, le cytomégavirus et le toxoplasme, ainsi que les herbicides à base de glyphosate (Roundal, Glyfor, Tornado, etc.). La liste des facteurs iatrogènes comprend les médicaments contre l'acné et la séborrhée contenant de l'acide 13-cis-rétinoïque (isotrétinoïne, Accutane); l'anticonvulsivant phénytoïne (Dilantin, Epanutin); les psychotropes diazépam, valium, relanium, seduxen.
Symptômes Syndrome de Treacher Collins
Dans la plupart des cas, les signes cliniques de la dysostose mandibulofasciale et leur degré d'expression dépendent des caractéristiques de la manifestation des mutations génétiques. Les premiers signes de cette anomalie sont généralement visibles chez l'enfant immédiatement après la naissance: le visage atteint du syndrome de Treacher-Collins présente une apparence caractéristique. De plus, les anomalies morphologiques sont généralement bilatérales et symétriques.
Les symptômes les plus évidents du syndrome de Treacher Collins sont:
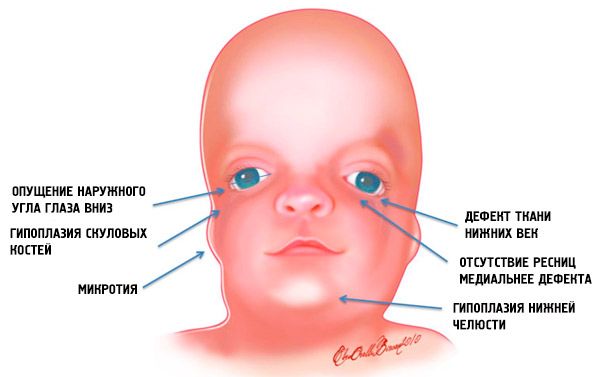
- sous-développement (hypoplasie) des os faciaux du crâne: zygomatiques, processus zygomatiques de l'os frontal, plaques ptérygoïdiennes latérales, sinus paranasaux, mâchoire inférieure et protubérances des épiphyses osseuses (condyles);
- sous-développement des os de la mâchoire inférieure (micrognathie) et angle mandibulaire plus obtus que d'habitude;
- le nez est de taille normale, mais paraît grand en raison de l'hypoplasie des arcades sourcilières et du sous-développement ou de l'absence des arcades zygomatiques dans la région temporale;
- les fentes oculaires sont vers le bas, c'est-à-dire que la forme des yeux est anormale, avec les coins extérieurs tombants vers le bas;
- défauts des paupières inférieures (colobome) et absence partielle de cils sur celles-ci;
- oreillettes de forme irrégulière présentant une large gamme de déviations, notamment leur emplacement dans le coin de la mâchoire inférieure, l'absence de lobes, des fistules aveugles entre le tragus de l'oreille et le coin de la bouche, etc.
- rétrécissement ou fermeture (atrésie) du conduit auditif externe et anomalies des osselets de l'oreille moyenne;
- absence ou hypoplasie des glandes salivaires parotides;
- hypoplasie pharyngée (rétrécissement du pharynx et des voies respiratoires);
- non-fusion du palais dur (fente palatine), ainsi que l'absence, le raccourcissement ou l'immobilité du palais mou.
Ces anomalies anatomiques entraînent systématiquement des complications. Il s'agit de troubles auditifs fonctionnels tels qu'une surdité de transmission ou une surdité complète; d'une déficience visuelle due à une mauvaise formation des globes oculaires; de malformations du palais entraînant des difficultés d'alimentation et de déglutition. Il existe des troubles de l'occlusion dentaire (malocclusion) associés à des malformations de la mâchoire, qui, à leur tour, entraînent des problèmes de mastication et d'articulation. Les pathologies du voile du palais expliquent la voix nasale.
Complications et conséquences
Les conséquences des anomalies maxillo-faciales dans le syndrome de Treacher Collins sont qu'à la naissance, les capacités intellectuelles de l'enfant sont normales, mais en raison de défauts auditifs et d'autres troubles, un retard mental secondaire est observé.
De plus, les enfants présentant de tels défauts ressentent profondément leur infériorité et souffrent, ce qui affecte négativement leur système nerveux et leur psychisme.
Diagnostics Syndrome de Treacher Collins
Le diagnostic postnatal du syndrome de Treacher Collins repose essentiellement sur les signes cliniques. La dysostose craniofaciale est facilement identifiée lorsque le syndrome est pleinement exprimé, mais lorsque les symptômes pathologiques sont peu exprimés, le diagnostic peut être difficile à établir.
Dans ce cas, une attention particulière doit être portée à l'évaluation de toutes les fonctions associées aux anomalies, notamment celles affectant la respiration (en raison du risque d'apnée du sommeil). L'efficacité de l'alimentation et la saturation en oxygène de l'hémoglobine doivent également être évaluées et surveillées.
Plus tard, le 5e ou 6e jour après la naissance, l'étendue des dommages auditifs devra être déterminée à l'aide de tests audiologiques, qui devront être effectués à la maternité.
Un examen est prescrit, au cours duquel des diagnostics instrumentaux sont effectués par fluoroscopie de dysmorphologie craniofaciale; pantomographie (radiographie panoramique des structures osseuses du crâne facial); tomodensitométrie crânienne complète dans diverses projections; TDM ou IRM du cerveau pour déterminer l'état du conduit auditif interne.
Le diagnostic prénatal le plus précoce des anomalies maxillo-faciales en présence du syndrome de Treacher Collins dans les antécédents familiaux est possible par biopsie des villosités choriales à 10-11 semaines de grossesse (la procédure menace de fausse couche et d'infection de l'utérus).
Des analyses de sang sont également effectuées auprès des membres de la famille; à 16-17 semaines de grossesse, le liquide amniotique est analysé (amniocentèse transabdominale); à 18-20 semaines de grossesse, une fœtoscopie est réalisée et du sang est prélevé dans les vaisseaux fœtaux du placenta.
Mais le plus souvent, l'échographie est utilisée dans le diagnostic prénatal de ce syndrome chez le fœtus (à 20-24 semaines de grossesse).
Quels tests sont nécessaires?
Diagnostic différentiel
Ces mêmes méthodes sont utilisées par les spécialistes lorsque des diagnostics différentiels sont nécessaires pour reconnaître le syndrome léger de Treacher Collins et le distinguer d'autres anomalies congénitales des os cranio-faciaux, en particulier: les syndromes d'Apert, de Crouzon, de Nager, de Peters-Hewels, de Hellermann-Steph, ainsi que la microsomie hémifaciale (syndrome de Goldenhar), l'hypertélorisme, la fusion prématurée des sutures crâniennes (craniosynostose) ou la fusion altérée des os du visage (craniosynostose).
Qui contacter?
Traitement Syndrome de Treacher Collins
Comme dans tous les cas de malformations congénitales d'origine génétique, le traitement des formes sévères du syndrome de Treacher Collins est exclusivement palliatif, car il n'existe tout simplement pas de traitement pour ces pathologies. Le spectre et l'intensité des déformations de ce syndrome sont vastes et, par conséquent, la nature et l'intensité de l'intervention médicale offrent de nombreuses options.
Les appareils auditifs sont utilisés pour corriger et améliorer l’audition, et les séances d’orthophonie sont utilisées pour améliorer la parole.
Des interventions chirurgicales sont nécessaires dès le plus jeune âge dans les cas graves de rétrécissement des voies respiratoires (trachéotomie) et du larynx (gastrostomie pour l'alimentation). Une correction chirurgicale du palais peut également être nécessaire.
Les chirurgies d'allongement mandibulaire sont pratiquées à l'âge de 2-3 ans ou plus. La reconstruction des tissus mous comprend la correction du colobome de la paupière inférieure et la chirurgie plastique auriculaire.
La prévention
La prévention du syndrome de Treacher Collins implique que les futurs parents consultent en conseil génétique, et s'il existe des antécédents familiaux du syndrome, la question de la possibilité d'une grossesse elle-même se pose - afin d'éviter la naissance d'un enfant présentant des anomalies craniofaciales.
Prévoir
Quel est le pronostic de cette pathologie? Il dépend du degré de déformation et de l'intensité des symptômes. Le syndrome de Treacher-Collins est un diagnostic permanent.
 [ 25 ]
[ 25 ]