Expert médical de l'article

Nouvelles publications
Syndrome d'Aper
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.

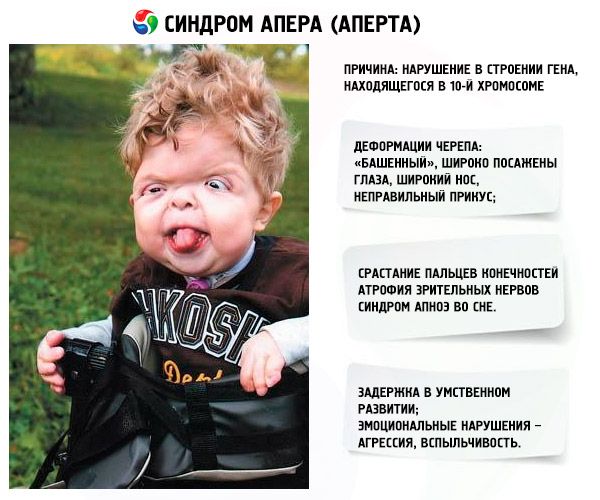
Le syndrome d'Apert est une pathologie génétique qui se traduit par des défauts de développement du crâne (yeux écartés, crâne excessivement haut), et en plus de cela, des membres inférieurs et supérieurs (les doigts sont complètement fusionnés; des doigts supplémentaires peuvent également apparaître).
 [
[ Causes Syndrome d'Aper
Le développement du syndrome est provoqué par un trouble de la structure d'un gène situé sur le chromosome 10. Ce gène est responsable du processus de séparation des doigts et, en outre, de la fermeture rapide des sutures crâniennes.
De plus, les maladies infectieuses contractées pendant la grossesse (comme la rubéole, la tuberculose, la syphilis, la grippe et la méningite) et l'exposition de la mère aux rayons X sont considérées comme les causes de cette pathologie. Le plus souvent, ce syndrome est détecté chez les enfants nés de parents âgés.
Pathogénèse
Le syndrome d'Apert est héréditaire. Son type est autosomique dominant (c'est-à-dire que dans une famille où l'un des futurs parents est malade, la probabilité d'avoir un enfant atteint de cette maladie est de 50 à 100 %).
Une mutation unique du récepteur 2 du facteur de croissance des fibroblastes (FGFR2) entraîne une augmentation du nombre de cellules progénitrices qui se développent le long de la voie ostéogénique. Ceci entraîne une augmentation de la formation de matrice osseuse sous-périostée et une ossification prématurée des sutures crâniennes pendant le développement fœtal. L'ordre et la vitesse de fusion des sutures déterminent le degré de déformation et d'invalidité. Une fois le matériel de suture cicatrisé, la croissance des autres tissus perpendiculaires à cette suture est limitée, et les os fusionnés forment un seul et même échafaudage osseux.
La première preuve génétique que la syndactylie dans le syndrome d'Apert est due à un défaut du récepteur du facteur de croissance des kératinocytes (KGFR) a été l'observation d'une corrélation entre l'expression du KGFR dans les fibroblastes et la gravité de la syndactylie.
L'amblyopie et le strabisme sont plus fréquents chez les patients porteurs de la mutation FGFR2 Ser252Trp, et l'atrophie papillaire est plus fréquente chez les patients porteurs de la mutation FGFR2 Pro253Arg. Les patients porteurs de la mutation FGR2 Ser252Trp présentent une prévalence de déficience visuelle significativement plus élevée que ceux porteurs de la mutation FGFR2 Pro253Arg.
Symptômes Syndrome d'Aper
Certaines manifestations de la maladie sont clairement visibles dès la naissance du bébé, car elles se développent dans l'utérus. Parmi les principaux symptômes du syndrome:
- Le crâne est déformé - il s'étire en hauteur, acquérant une apparence de « tour »; de plus, les yeux sont écartés et légèrement bombés (car la taille des orbites diminue); le nez s'élargit et une morsure incorrecte se forme (les dents supérieures dépassent excessivement);
- Les doigts des membres sont complètement fusionnés (principalement l'annulaire, ainsi que le majeur et l'index) et ressemblent à une membrane cutanée ou à une fusion complète des os; de plus, des doigts supplémentaires peuvent pousser;
- Retard mental (ne survient pas chez tout le monde);
- Les nerfs optiques s'atrophient, ce qui entraîne une diminution de l'acuité visuelle (dans certains cas, une perte complète de la vision se développe);
- L'augmentation de la pression intracrânienne, qui survient à la suite d'une fermeture prématurée des sutures crâniennes, se manifeste sous la forme de maux de tête et de vomissements avec nausées;
- Comme la mâchoire supérieure reste sous-développée, des problèmes respiratoires surviennent;
- Le syndrome d’apnée du sommeil est un phénomène courant.
- Manifestations émotionnelles – agressivité, manque de retenue, forte irascibilité.
Complications et conséquences
La plupart des patients présentent un certain degré d'obstruction des voies aériennes supérieures dès la petite enfance. Les voies aériennes supérieures sont difficilement perméables en raison de la taille réduite du nasopharynx et des choanes, et les voies aériennes inférieures peuvent entraîner un décès prématuré dû à des anomalies du cartilage trachéal.
Diagnostics Syndrome d'Aper
Pour établir un diagnostic, les étapes suivantes sont nécessaires:
- Le médecin doit analyser les symptômes du patient ainsi que ses antécédents médicaux. Il est nécessaire de rechercher d'éventuels cas de pathologies similaires dans la famille.
- Examen neurologique pour évaluer la forme du crâne et le développement intellectuel du patient (questionnaires spéciaux ainsi qu'entretien);
- Examen du fond d'œil pour détecter la présence de symptômes d'augmentation de la pression intracrânienne (gonflement du disque optique, ainsi que flou de ses bords);
- Pour évaluer l’état du crâne, une radiographie est réalisée;
- Imagerie par ordinateur et par résonance magnétique de la tête pour examiner la structure du cerveau et du crâne couche par couche, pour déterminer la présence de symptômes de fusion prématurée des sutures crâniennes, et en plus, d'hydrocéphalie (en raison de l'augmentation de la pression intracrânienne, un excès de liquide céphalo-rachidien s'accumule (c'est le liquide céphalo-rachidien qui favorise le processus métabolique, ainsi que la nutrition du cerveau));
- Radiographie des pieds et des mains pour déterminer la cause de la fusion des doigts (ceci est important pour planifier une intervention chirurgicale ultérieure);
- Une consultation avec un généticien médical et un neurochirurgien peut être prescrite.
Tests
Une analyse génétique des mutations courantes survenant dans le gène de type FGFR2 est en cours.
Diagnostic différentiel
Ce syndrome doit être différencié des autres pathologies génétiques associées à une craniosténose, telles que les syndromes de Pfeiffer, de Crouzon, de Saethre-Chotzen et de Carpenter. Des tests génétiques moléculaires permettent d'exclure ces anomalies.
Qui contacter?
Traitement Syndrome d'Aper
La chirurgie est considérée comme le seul traitement efficace du syndrome d’Apert: elle permet de corriger les défauts physiques individuels et corrige également le retard mental.
Au cours de cette intervention, la suture coronale est fermée afin de prévenir toute lésion cérébrale. La technique la plus courante est la distraction cranio-faciale, qui implique une traction crânienne graduée. Une chirurgie orthodontique et/ou orthognathique est pratiquée pour corriger les défauts faciaux individuels.
De plus, les patients subissent une correction chirurgicale de la fusion des doigts.
Références
Génétique médicale. Guide national. Edité par Ginter EK, Puzyrev VP GEOTAR-Media. 2022