Expert médical de l'article

Nouvelles publications
Le syndrome de Pierre Robin
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.

Le syndrome de Pierre Robin, également connu en médecine sous le nom d'anomalie de Robin, est une pathologie congénitale du développement de la mâchoire. La maladie doit son nom au dentiste français P. Robin, qui en a décrit tous les signes. Lannelongue et Ménard ont décrit le syndrome de Pierre Robin pour la première fois en 1891 dans leur rapport sur deux patients atteints de micrognathie, de fente palatine et de rétroglossoptose. En 1926, Pierre-Robin a publié un cas de la maladie chez un nourrisson présentant les signes du syndrome classique. Jusqu'en 1974, cette triade de signes était connue sous le nom de syndrome de Robin-Pierre. Cependant, ce syndrome est désormais utilisé pour décrire des malformations avec présence simultanée de multiples anomalies.
Épidémiologie
Il s'agit d'une anomalie congénitale hétérogène dont la prévalence est de 1 pour 8 500 naissances vivantes. Le ratio homme/femme est de 1:1, sauf pour la forme liée à l'X.
Parmi ces patients, 50 % des nourrissons présentent une fente palatine incomplète, les autres naissent avec un palais arqué et anormalement haut, mais sans fente.
Causes Le syndrome de Pierre Robin
La possibilité d'une transmission autosomique récessive de la maladie est envisagée. Il existe deux types de syndrome selon l'étiologie: isolé et génétiquement déterminé. Le type isolé se développe suite à une compression de la partie inférieure de la mâchoire pendant le développement embryonnaire. La compression peut se développer suite à:
- La présence de scellements localisés dans l'utérus (kystes, cicatrices, tumeurs).
- Grossesse multiple.
De plus, le développement de la mâchoire du fœtus peut être perturbé par:
- Infections virales dont la future mère a souffert pendant sa grossesse.
- Troubles neurotrophiques.
- Quantité insuffisante d'acide folique dans le corps d'une femme enceinte.
Pathogénèse
Le syndrome de Pierre Robin est causé par des troubles embryonnaires causés par une grande variété de pathologies au cours de la période prénatale.
Il existe trois théories physiopathologiques pouvant expliquer la survenue du syndrome de Pierre Robin.
Théorie mécanique: Cette théorie est la plus probable. Le sous-développement de l'appareil mandibulaire survient entre la 7e et la 11e semaine de grossesse. La position haute de la langue dans la cavité buccale entraîne la formation de fentes palatines, empêchant la fermeture de la veine cave. Cette théorie explique la fente classique en U inversé et l'absence de fente labiale associée. L'oligohydramnios pourrait jouer un rôle dans l'étiologie, car l'absence de liquide amniotique peut entraîner une déformation du menton et une compression ultérieure de la langue entre les veines caves.
Théorie neurologique: Un retard du développement neurologique a été noté avec l'électromyographie des muscles de la luette et des colonnes pharyngées, et du goût en raison d'un retard de conduction dans le nerf hypoglosse.
Théorie de la dysneurorégulation du rhombencéphale: Cette théorie est basée sur la perturbation du développement du rhombencéphale au cours de l'ontogenèse.
Le développement insuffisant de la partie inférieure de la mâchoire de l'enfant entraîne une réduction significative de la cavité buccale. Ceci provoque une pseudomacroglossie, c'est-à-dire un déplacement de la langue vers l'arrière de la paroi pharyngée. Cette pathologie entraîne une obstruction des voies respiratoires.
Tant que le bébé pleure ou bouge, les voies respiratoires restent dégagées, mais dès que le bébé s'endort, l'obstruction se produit à nouveau.
En raison de troubles respiratoires, l'alimentation du bébé est très difficile. À ce stade, une obstruction des voies respiratoires survient presque systématiquement. Sans correction médicale, une telle pathologie peut entraîner un épuisement grave de l'organisme, voire la mort.
Symptômes Le syndrome de Pierre Robin
La maladie se caractérise par trois symptômes principaux:
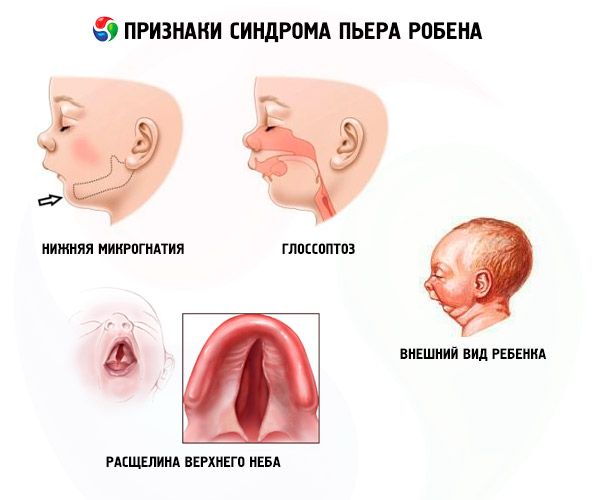
- La micrognathie inférieure (sous-développement de la mâchoire inférieure, observée dans 91,7 % des cas) se caractérise par une rétraction de l'arcade dentaire inférieure de 10 à 12 mm en arrière de l'arcade supérieure. La mâchoire inférieure présente un petit corps et un angle obtus. L'enfant atteint un développement normal vers 5-6 ans.
- Glossoptose (rétraction de la langue due à son développement insuffisant, observée dans 70 à 85 % des cas).
- La macroglossie et l’ankyloglossie sont des symptômes relativement rares, observés dans 10 à 15 % des cas.
- Une fissure apparaît dans le ciel.
- Bradypnée et dyspnée.
- Cyanose légère.
- L'asphyxie, qui survient le plus souvent lors des tentatives d'alimentation du bébé.
- La déglutition est impossible ou très difficile.
- Envie de vomir.
- Anomalies auriculaires dans 75% des cas.
- Une perte auditive conductrice survient chez 60 % des patients, tandis qu'une atrésie du conduit auditif externe survient chez seulement 5 % des patients, une pneumatisation insuffisante de la cavité mastoïde de l'os temporal.
- Anomalies de l'oreille interne (aplasie des canaux semi-circulaires latéraux, grand aqueduc vestibulaire, perte des cellules ciliées cochléaires).
- Les malformations nasales sont rares et consistent principalement en des anomalies de la racine nasale.
- Des malformations dentaires surviennent dans 30 % des cas. Une laryngomalacie et une insuffisance vélopharyngée surviennent chez environ 10 à 15 % des patients atteints du syndrome de Pierre Robin.
Caractéristiques systémiques du syndrome de Pierre Robin
Des anomalies systémiques du développement sont décrites dans 10 à 85 % des cas enregistrés.
Des anomalies oculaires surviennent chez 10 à 30 % des patients. Elles peuvent inclure: hypermétropie, myopie, astigmatisme, sclérose cornéenne et sténose du canal lacrymo-nasal.
Pathologies cardiovasculaires: souffles cardiaques bénins, sténose de l’artère pulmonaire, persistance du canal artériel, fenêtre ovale, communication interauriculaire et hypertension pulmonaire. Leur prévalence varie de 5 à 58 %.
Anomalies de l'appareil locomoteur (70 à 80 % des cas): syndactylie, dysplasie des phalanges, polydactylie, clinodactylie, hypermobilité articulaire et oligodactylie des membres supérieurs. Anomalies des membres inférieurs: anomalies du pied (pied bot, adduction métatarsienne), malformations fémorales (bassin valgus ou varus, fémurs courts), anomalies de la hanche (luxation congénitale, contractures), anomalies de l'articulation du genou (GENU VALGUS, synchondrose). Malformations de la colonne vertébrale: scoliose, cyphose, lordose, dysplasie vertébrale, agénésie du sacrum et du sinus coccygien.
Pathologie du système nerveux central: épilepsie, retard de développement du système nerveux, hydrocéphalie. La fréquence des anomalies du système nerveux central est d'environ 50 %.
Anomalies génito-urinaires: testicules non descendus (25 %), hydronéphrose (15 %) et hydrocèle (10 %).
Syndromes et affections associés: syndrome de Stickler, syndrome de trisomie 11q, trisomie 18, syndrome de délétion 4q, arthropathie rhumatoïde, hypochondroplasie, syndrome de Moebius.
Étapes
Il existe trois stades de gravité de la maladie, qui dépendent de l’état des voies respiratoires de l’enfant:
- Légère: problèmes mineurs d'alimentation, mais respiration aisée. Le traitement est effectué en ambulatoire.
- Modérée – La respiration est modérément difficile, l'alimentation de l'enfant est modérément difficile. Le traitement est effectué en milieu hospitalier.
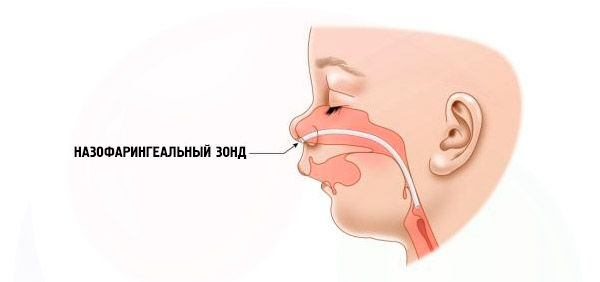
- Grave – la respiration est très difficile, l'enfant ne peut pas être nourri normalement. Il est nécessaire d'utiliser des dispositifs spéciaux (sonde intranasale).
Complications et conséquences
L'association de la micrognathie et de la glossoptose peut entraîner de graves complications respiratoires et des problèmes lors de l'alimentation de l'enfant.
Le syndrome de Pierre Robin entraîne les complications suivantes:
- Respiration stridente due à une obstruction des voies respiratoires. Laryngomalacie, voire asphyxie du sommeil.
- Le développement psychomoteur de l’enfant est très en retard par rapport à celui de ses pairs.
- Le développement physique est également en retard.
- La parole des patients est altérée.
- Infections fréquentes de l’oreille qui deviennent chroniques et entraînent une déficience auditive.
- Syndrome d'apnées obstructives du sommeil, la survenue de décès pendant le sommeil varie dans 14 à 91 % des cas.
- Problèmes de dents.
Diagnostics Le syndrome de Pierre Robin
Le diagnostic du syndrome de Pierre Robin est simple. Il repose sur les manifestations cliniques. Pour exclure d'autres pathologies, il est essentiel de consulter un généticien.
Les enfants atteints de l'anomalie congénitale de Robin présentent des difficultés respiratoires dès la naissance, la langue s'enfonçant constamment. Le bébé est agité, sa peau est bleutée et sa respiration sifflante sort de la poitrine à l'inspiration. Un étouffement peut survenir pendant la tétée. Le diagnostic peut également être posé par l'apparence inhabituelle de l'enfant, une « visage d'oiseau ». Souvent, les patients développent d'autres anomalies: myopie, cataracte, pathologie de l'appareil génito-urinaire, pathologie cardiaque, anomalies du développement de la colonne vertébrale.
Sur la base de ces manifestations cliniques, il ne sera pas difficile pour un spécialiste de poser un diagnostic correct.
Qui contacter?
Traitement Le syndrome de Pierre Robin
Le traitement est effectué immédiatement après la naissance d'un enfant atteint du syndrome de Pierre Robin. Si la maladie est légère, pour améliorer l'état du patient, il est nécessaire de maintenir constamment l'enfant en position verticale ou allongée sur le ventre. La tête du bébé doit être inclinée vers la poitrine. Pendant l'allaitement, il est déconseillé de maintenir l'enfant en position horizontale afin d'éviter tout contact des aliments avec les voies respiratoires.
Si le sous-développement de la mâchoire inférieure est très prononcé, une intervention chirurgicale est nécessaire pour ramener la langue rétractée à une position physiologique normale. Dans les cas graves, la langue est relevée et fixée sur la lèvre inférieure. Dans les cas très graves, une trachéotomie, une glossopexie et une ostéogenèse par distraction de la mâchoire inférieure doivent être réalisées.
Un traitement conservateur est également utilisé.
Médicaments
Phénobarbital. Ce somnifère et sédatif a un effet anticonvulsivant. Chaque comprimé contient 100 ml de phénobarbital. La posologie est individuelle, car elle dépend de la gravité de la maladie et de l'état de l'enfant. Ce médicament est déconseillé aux patients souffrant d'insuffisance hépatique, d'hyperkinésie, d'anémie, de myasthénie, de porphyrie, de diabète sucré, de dépression et d'intolérance aux composants. Les symptômes suivants peuvent survenir lors de sa prise: vertiges, asthénie, hallucinations, agranulocytose, nausées, hypotension artérielle et allergies.
Clonazépam. Médicament prescrit pour le traitement de l'épilepsie. Ce médicament contient le principe actif clonazépam, un dérivé de la benzodiazépine. Il possède des effets anticonvulsivants, anxiolytiques et myorelaxants. La dose est déterminée par le médecin traitant, mais ne doit pas dépasser la dose maximale de 250 µg par jour. Ne pas prendre en cas d'insomnie, d'hypertonie musculaire, d'agitation psychomotrice ou de troubles paniques. Les symptômes suivants peuvent survenir lors de la prise: léthargie, nausées, dysménorrhée, maux de tête, leucopénie, rétention urinaire ou incontinence, alopécie, allergie.
Sibazon. Disponible sous forme de solution et de comprimés rectaux. Le principe actif est un dérivé de benzodiazépine (sibazon). Il a un effet sédatif, anxiolytique et anticonvulsivant. La posologie est individuelle. Ce médicament est déconseillé aux patients souffrant d'hypercapnie chronique, de myasthénie ou d'intolérance aux benzodiazépines. L'utilisation de ce médicament peut entraîner les symptômes suivants: nausées, constipation, maux de tête, vertiges, hoquet, incontinence urinaire et allergies.
Lyophilisat de cortexine. Médicament à effet nootrope. Ce médicament contient un complexe de fractions polypeptidiques hydrosolubles et de glycine. La posologie est individuelle et prescrite par le médecin traitant en fonction de l'état du patient. Les patients présentant une intolérance à la cortexine sont déconseillés. Ce médicament peut provoquer des réactions allergiques.
Traitement de physiothérapie
En général, dans les stades légers du syndrome, on utilise une thérapie positionnelle, où l'enfant est placé sur le ventre en position verticale jusqu'à ce que la gravité force la mâchoire inférieure à se développer correctement.
Traitement chirurgical
Le traitement chirurgical est principalement utilisé pour corriger la glossoptose. Il existe plusieurs méthodes:
- Maintien de la langue par un fil d'argent. Le fil est passé à travers la partie inférieure de la gencive et la lèvre inférieure. Cette méthode est appelée Douglas.
- Méthode Duhamel: un fil d'argent épais est passé à la base de la langue et des deux joues du patient. À utiliser pendant trente jours maximum.
- Dispositifs orthopédiques pour l'extension et la fixation de la langue.
- À l’âge d’un an, une intervention chirurgicale pour corriger une fente palatine peut être réalisée.
Prévoir
Le pronostic et l'évolution de la maladie sont graves. Le plus souvent, le décès survient dans les premiers jours de vie, aux stades modéré et sévère de la maladie (la cause est l'asphyxie). De plus, le risque de décès au cours de la première année est assez élevé en raison des nombreuses infections.
Pour les patients de plus de deux ans, le pronostic est favorable.
 [ 36 ]
[ 36 ]