Expert médical de l'article

Nouvelles publications
Syndrome de Tolosa-Hunt
Dernière revue: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
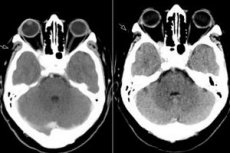
Syndrome de fissure orbitaire supérieure, ophtalmoplégie pathologique: tout cela n'est rien d'autre que le syndrome de Tolosa Hunt, une lésion des structures de la fissure orbitaire supérieure. Le processus touche généralement les vaisseaux orbitaires (artériels et veineux), les fibres nerveuses (nerfs oculomoteur, trochléaire, abducens, ainsi que la première branche du nerf trijumeau) et le sinus caverneux voisin. Cette maladie peut être classée comme une pathologie relativement rare et difficile à diagnostiquer. [ 1 ]
Épidémiologie
Le syndrome de Tolos Hunt a été décrit il y a environ 70 ans. Il a été étudié par le neurologue espagnol E. Tolos. Quelques années plus tard, ses travaux ont été complétés par ceux du médecin ophtalmologue anglais W. Hunt. Les noms des médecins-chercheurs ont servi de base à la dénomination du syndrome.
Le syndrome de Tolosa Hunt touche autant les hommes que les femmes. La pathologie est généralement unilatérale et s'observe aussi bien du côté gauche que du côté droit. Un syndrome bilatéral est possible, mais ne survient que dans des cas isolés.
L'âge moyen des personnes atteintes est de 50 ans. En général, le syndrome de Tolosa Hunt apparaît entre 15 et 85 ans. La plupart des patients sont âgés: le développement de la maladie est favorisé par de multiples troubles cardiovasculaires, ainsi que par des modifications tissulaires liées à l'âge.
Le symptôme le plus courant de la maladie est la manifestation d'une crise migraineuse classique: la personne ressent une céphalée soudaine, pulsatile et unilatérale, avec une irradiation de l'orbite. Le syndrome de Tolosa Hunt étant dépourvu de symptômes spécifiques typiques, cette pathologie est souvent qualifiée de « caméléon neurologique »: son diagnostic est complexe et nécessite de la différencier de nombreuses autres maladies.
Les patients atteints du syndrome de Tolosa Hunt sont observés périodiquement dans différents pays du monde, sans particularité territoriale ni saisonnière. Le taux d'incidence est de 0,3 à 1,5 cas pour 1 million d'habitants. [ 2 ]
Causes Syndrome de Tolosa-Hunt
Au cours de l'enquête sur les causes du développement du syndrome de Tolosa Hunt, les scientifiques ont découvert les faits suivants:
- dans la plupart des cas, la maladie a été provoquée par une inflammation immunitaire de la paroi externe du sinus caverneux;
- dans certains cas, les causes étaient des malformations vasculaires, des processus tumoraux dans le cerveau (formes primaires et secondaires), une pachyméningite crânienne localisée, une myosite orbitaire, une périartérite noueuse et la formation de thrombus dans le sinus caverneux;
- Chez environ 30 % des patients, la cause du trouble ne peut être déterminée, le diagnostic de syndrome de Tolosa Hunt idiopathique a donc été établi.
Considérons ces prétendues raisons plus en détail.
- Le développement auto-immun du syndrome est associé à la fois à l'hypothermie et à des pathologies infectieuses récentes, ainsi qu'à un stress profond. La forme auto-immune de la maladie se caractérise par: une apparition brutale, une évolution récurrente et une efficacité élevée de la corticothérapie. Cette forme de la maladie touche le plus souvent les hommes.
- Les malformations vasculaires surviennent souvent en cas d'hypertension artérielle décompensée. Les femmes sont plus souvent touchées. La maladie débute de manière aiguë, la douleur est modérée, pratiquement sans exophtalmie ni chémosis.
- Parmi les processus tumoraux susceptibles de conduire au développement du syndrome de Tolosa Hunt, les plus fréquents étaient les tumeurs cérébrales primaires, les tumeurs métastatiques avec des foyers primaires dans les poumons, les bronches, la prostate ou les métastases de mélanome cutané.
- La pachyméningite crânienne localisée entraîne un syndrome d'apparition aiguë en l'absence de signes cérébraux et méningés généraux, sans exophtalmie. Le diagnostic est confirmé morphologiquement par biopsie.
- La myosite orbitaire provoque un début subaigu, avec une douleur intense et une exophtalmie, un chémosis prononcé et une vision double.
- La thrombose du sinus caverneux entraîne une ophtalmoplégie totale. Le diagnostic est confirmé par imagerie par résonance magnétique.
- La périartérite nodulaire peut provoquer le développement du syndrome de Tolosa Hunt plusieurs mois après le début de la maladie.
Dans la plupart des cas, le mécanisme auto-immun est à l'origine de la pathologie, comme l'ont démontré de nombreux spécialistes. Le caractère auto-immun est notamment indiqué par les facteurs suivants:
- cours récurrent;
- troubles dysmuniques;
- dissociation des protéines et des cellules dans le liquide céphalo-rachidien et augmentation des niveaux de cytokines pro-inflammatoires dans le liquide céphalo-rachidien et le sérum sanguin. [ 3 ]
Facteurs de risque
Les scientifiques n'ont pas encore déterminé la cause exacte du syndrome de Tolosa Hunt. Ils ont toutefois réussi à identifier certains facteurs influençant son développement:
- Prédisposition génétique aux maladies auto-immunes en général. Si l'un des membres de la famille a souffert ou souffre d'une maladie auto-immune, d'autres membres de la famille peuvent également présenter des pathologies similaires ou d'autres pathologies avec un mécanisme de développement similaire. Ce facteur reste une hypothèse qui nécessite des recherches et des preuves supplémentaires.
- Facteurs environnementaux, notamment les habitudes alimentaires, les conditions environnementales, la qualité de l’eau, les risques industriels, etc.
- Situations de stress intense, stress fréquent et chocs psycho-émotionnels, changements hormonaux puissants (notamment grossesse, ménopause, etc.).
- Maladies infectieuses chroniques à long terme, notamment l’hépatite, l’infection par l’herpèsvirus, le cytomégalovirus, etc.
- Hypothermie, radiations, autres irritants puissants et facteurs nocifs.
Pathogénèse
Le mécanisme étiologique du développement du syndrome de Tolosa-Hunt n'a pas été entièrement élucidé. Le rôle déterminant est attribué aux réactions auto-immunes. De nombreux scientifiques supposent que les infections virales et microbiennes, les situations stressantes et les radiations ne constituent que des facteurs déclenchants. Il n'existe aucune preuve solide d'un lien entre l'entrée de micro-organismes pathogènes dans l'organisme et le développement du syndrome de Tolosa-Hunt. Cependant, on soupçonne l'implication du cytomégalovirus dans le processus auto-immun, qui contribue à la formation de granulomes. [ 4 ]
Le schéma pathogénique est dû à l'apparition d'un processus inflammatoire granulomateux local au niveau de la paroi externe du sinus caverneux, de la section infraclinoïde ou supraclinoïde de l'artère carotide interne, entraînant son rétrécissement. Un rôle important est également joué par le trouble de la protection immunitaire humorale et cellulaire. Le versant humoral du syndrome est associé à une production accrue d'anticorps cytoplasmiques antineutrophiles agissant contre les enzymes protéinase-3, myéloperoxydase et une protéine membranaire spécifique capable de lier les endotoxines. Vraisemblablement, les anticorps cytoplasmiques stimulent les neutrophiles existants, ce qui les amène à attaquer les organes cibles; le processus inflammatoire se développe notamment au niveau de la paroi externe du sinus caverneux.
Les modifications cellulaires jouent également un rôle dans le développement du syndrome de Tolosa Hunt, comme le démontre la prédominance des lymphocytes T, des macrophages et des plasmocytes dans les granulomes.
Il existe des informations sur des structures endothéliales hautement actives et des cytokines anti-inflammatoires, ce qui indique une tendance du processus pathologique à devenir chronique.
Dans des cas isolés, des modifications nécrotiques focales ont été observées dans la zone de la paroi externe du sinus caverneux.
Symptômes Syndrome de Tolosa-Hunt
Les symptômes caractéristiques du syndrome de Tolosa Hunt apparaissent soudainement et de manière inattendue. Les principaux symptômes sont les suivants:
- Douleur intense dans la région de l'orbite oculaire, extrêmement désagréable, lancinante, se propageant de la région frontale aux arcades sourcilières, aux yeux et plus loin dans toute la tête.
- Vision double, détectée après l'apparition de la douleur. Il devient alors extrêmement difficile pour la personne de se concentrer et d'examiner un objet.
- Le trouble de la fonction motrice du globe oculaire, appelé ophtalmoplégie, est principalement unilatéral. Il peut se manifester à des degrés variables, selon la gravité du processus pathologique et l'étendue de la lésion.
- Œdème conjonctival.
- Déplacement antérieur du globe oculaire (exophtalmie, yeux « exorbités »).
- Déviation de l'axe visuel d'un globe oculaire vers le côté, strabisme, qui est typique d'une lésion nerveuse unilatérale.
- Détérioration générale de l'état de santé, légère augmentation de la température, faiblesse, irritabilité.
Le tableau clinique évolue progressivement, les symptômes changent et s'aggravent, mais peuvent disparaître aussi soudainement qu'ils sont apparus. Cependant, en l'absence de traitement approprié, le syndrome de Tolosa Hunt réapparaît avec des rechutes.
Les symptômes neurologiques sont causés par la localisation locale du processus douloureux. La douleur résulte de l'irritation de la première branche du nerf trijumeau, qui passe près du tronc du nerf oculomoteur, et se manifeste au niveau de l'orbite, du front, des tempes et de la base du nez. L'intensité de la douleur varie de modérée à intense.
Des symptômes atypiques sont possibles, caractérisés par l'absence de douleur. Ceci peut être observé lorsque la lésion est localisée avant que la cinquième paire ne pénètre dans le sinus caverneux.
Les troubles oculomoteurs se manifestent généralement par une vision double lors du regard direct.
Si le processus douloureux est localisé au niveau de l'apex orbitaire, des manifestations neurologiques sont souvent associées à des troubles de l'analyseur visuel. Il en résulte un œdème ou une atrophie du disque optique, et un scotome central est souvent observé. Une exophtalmie (yeux exorbités) et un chémosis (œdème conjonctival) sont possibles, causés par des modifications infiltrantes du tissu rétrobulbaire et des difficultés d'écoulement veineux depuis l'orbite.
Premiers signes
Le syndrome de Tolosa Hunt n'ayant pas encore été suffisamment étudié, les scientifiques continuent d'élucider les mécanismes possibles de son développement. Compte tenu des critères définis par la Société internationale de neurologie, le diagnostic de syndrome de Tolosa Hunt est justifié par la présence d'un granulome de la paroi externe du sinus caverneux, détecté lors d'une IRM cérébrale ou d'une biopsie.
La liste des signes acceptés comme critères diagnostiques du syndrome est la suivante:
- douleur « cueillante » ou « tordue » dans une orbite oculaire avec développement ultérieur d'une paralysie musculaire (ophtalmoplégie);
- lésions combinées des nerfs oculomoteurs, de la première branche du nerf trijumeau et du plexus nerveux périartériel;
- une aggravation du tableau clinique sur plusieurs jours (ou en 1 à 2 semaines);
- la possibilité de rémission spontanée (dans certains cas – avec préservation résiduelle des défauts);
- la probabilité d’une rechute du syndrome, des mois ou des années plus tard;
- tableau systémique inchangé, aucune lésion en dehors du sinus carotidien;
- la présence d'un effet positif de la corticothérapie.
Il existe une autre liste diagnostique similaire de caractéristiques proposée en 2003. Selon cette liste, le syndrome de Tolosa Hunt est considéré comme le résultat de la prolifération de tissu granulomateux dans le sinus caverneux, la fissure orbitaire supérieure et la cavité orbitaire:
- un ou plusieurs épisodes de douleur unilatérale dans la région orbitaire qui disparaissent sans traitement pendant quelques semaines;
- atteinte du nerf crânien (III, IV ou VI) sous forme de parésie, présence d'un granulome confirmée par imagerie par résonance magnétique ou biopsie;
- l'apparition d'une parésie simultanément au syndrome douloureux, ou dans les 14 jours qui suivent celui-ci;
- disparition de la parésie et du syndrome douloureux dans les 3 jours suivant le début de la corticothérapie.
Formes
Dans le syndrome de Tolosa Hunt, les côtés gauche et droit sont affectés avec une fréquence à peu près égale, de sorte que la pathologie est divisée en côté gauche ou côté droit.
La maladie est généralement unilatérale. Des lésions bilatérales n'ont été observées que dans de très rares cas.
Le tableau clinique de la maladie peut évoluer selon les étapes suivantes:
- aiguë ou subaiguë, qui survient après une maladie infectieuse virale récente, une hypothermie, une forte augmentation de la pression artérielle, parfois sans raison évidente;
- maladie chronique récidivante, avec une augmentation progressive des symptômes et des exacerbations périodiques.
De plus, le syndrome de Tolosa Hunt peut être:
- totale, avec atteinte de tous les nerfs qui passent par la fissure orbitaire supérieure;
- incomplet, avec atteinte dans le processus pathologique des nerfs VI, IV, III paires et de la branche I de la paire V dans diverses combinaisons.
En ce qui concerne le sinus, on peut distinguer les formes antérieure, moyenne et postérieure du syndrome de Tolosa Hunt.
Complications et conséquences
Le syndrome de Tolosa Hunt s'accompagne de douleurs intenses, entraînant une perte de sommeil et des troubles émotionnels et mentaux. Les personnes malades deviennent irritables et émotionnellement instables. En l'absence de traitement approprié, des troubles névrotiques peuvent apparaître: états dépressifs, neurasthénie et hypocondrie se développent. La capacité de travail est considérablement réduite et le patient devient renfermé.
Le syndrome de Tolosa Hunt se caractérise par une évolution récidivante, fréquente dans les maladies auto-immunes. La durée de la rémission peut être très variable: la durée maximale d'asymptomatique enregistrée était de 11 ans. Après traitement, le risque de rechutes est significativement réduit. Si des exacerbations surviennent, elles sont moins graves.
Diagnostics Syndrome de Tolosa-Hunt
Il est souvent difficile pour les médecins de diagnostiquer immédiatement le syndrome de Tolosa Hunt, car les symptômes sont très similaires à ceux d'autres maladies plus courantes. Dans la plupart des cas, une consultation complémentaire auprès de plusieurs spécialistes est nécessaire: neurologue, ophtalmologue, endocrinologue, oncologue, neurochirurgien, etc.
Au premier stade, il est nécessaire d’exclure les maladies malignes, les anévrismes, la méningite, etc.
Le plus souvent, le diagnostic du syndrome de Tolosa Hunt est posé par exclusion: le patient subit une série de tests pour écarter d'autres pathologies probables. Les examens suivants sont nécessaires:
- image sanguine détaillée;
- étude de la fonction hormonale de la glande thyroïde;
- étude du taux de protéines totales dans le sang (pour évaluer la qualité du métabolisme des protéines);
- analyse du liquide céphalo-rachidien.
- Le diagnostic instrumental implique la réalisation des procédures de diagnostic suivantes:
- imagerie par résonance magnétique du cerveau et de la région orbitaire, avec et sans contraste;
- angiographie par résonance magnétique;
- angiographie par soustraction numérique (angiographie par soustraction intraveineuse);
- Tomodensitométrie cérébrale et orbitaire avec et sans contraste.
L'IRM avec injection de gadolinium est la modalité de choix pour l'évaluation du syndrome de Stevens-Johnson (STH) et peut mettre en évidence une hypertrophie et un rehaussement anormaux du SC, s'étendant de la fissure orbitaire supérieure à l'apex orbitaire. Les résultats d'IRM rapportés sur les images pondérées en T1 et T2 sont extrêmement variables et non spécifiques. L'IRM joue un rôle clé dans le diagnostic et permet d'exclure d'autres lésions fréquentes associées au SC, évitant ainsi le recours à des procédures invasives à haut risque telles que la biopsie du SC, seule méthode permettant d'obtenir une confirmation histopathologique de cette maladie.[ 5 ]
Ces études permettent d'identifier des traces de processus inflammatoires dans le sinus caverneux, la fissure orbitaire supérieure ou l'apex orbitaire. Des traces d'inflammation dans la région orbitaire sur des coupes transversales, en l'absence de paralysie des nerfs crâniens, sont considérées comme plus bénignes en termes de pronostic.
Il est conseillé à certains patients suspectés d’être atteints du syndrome de Tolosa Hunt de subir une biopsie pour écarter un cancer.
Diagnostic différentiel
La pratique clinique indique que des symptômes similaires peuvent être présents dans de nombreuses pathologies somatiques et neurologiques:
- dans les processus inflammatoires microbiens, viraux et fongiques affectant les méninges ou la paroi externe du sinus caverneux;
- dans les processus tumoraux du cerveau et de l'orbite - par exemple, dans l'adénome hypophysaire, le craniopharyngiome, le neurinome, le méningiome de l'aile du sphénoïde, dans les métastases cérébrales ou orbitaires;
- dans les malformations vasculaires - en particulier dans les anévrismes veineux-artériels, les fistules carotido-caverneuses, etc., ainsi que dans les dissections de branches de l'artère carotide interne;
- pour thrombose, formations kystiques du sinus caverneux, lymphome;
- pour la sarcoïdose, la myosite orbitaire (muscles oculaires), la granulomatose de Wegener (granulomatose avec polyangéite), l'ophtalmomigraine et certaines pathologies sanguines.
Le diagnostic différentiel consiste à déterminer la possibilité de développer toutes les maladies ci-dessus, sur la base des résultats d'une enquête, d'un examen, d'études en laboratoire et instrumentales.
Le plus souvent, le syndrome de Tolosa Hunt doit être distingué des pathologies suivantes:
- blocage du sinus caverneux par un thrombus;
- Syndrome de Rochon-Duvignod;
- syndrome de l'espace rétrosphénoïdal (syndrome de Jacot);
- syndrome de Raeder paratrigéminal;
- polyneuropathie crânienne.
Qui contacter?
Traitement Syndrome de Tolosa-Hunt
Le syndrome de Tolosa Hunt répond bien au traitement immunosuppresseur par corticostéroïdes. Ces médicaments sont capables de supprimer la réponse immunitaire agressive et ses effets néfastes sur les tissus de l'organisme.
Les médicaments les plus couramment prescrits sont la prednisolone, la méthylprednisolone, la cortisone ou d'autres médicaments ayant montré des effets positifs dans le traitement de pathologies auto-immunes connues. Les bénéfices des stéroïdes sont probablement liés à leur mécanisme antioxydant et/ou à la capacité de doses aussi élevées à réduire l'œdème et l'ischémie subséquente dans les zones affectées. [ 6 ]
Outre les corticostéroïdes, il est conseillé d'utiliser des analgésiques et des anticonvulsivants. Des préparations multivitaminées complexes sont indispensables.
En suivant scrupuleusement toutes les instructions et recommandations de votre médecin, les symptômes douloureux du syndrome de Tolosa Hunt disparaissent rapidement: les patients constatent une nette amélioration de leur bien-être dès le deuxième ou le troisième jour. Dans la grande majorité des cas, la capacité de travail est maintenue. [ 7 ]
Les dosages optimaux et la fréquence de prise des médicaments hormonaux sont déterminés individuellement. Il n'existe pas de schéma thérapeutique universellement reconnu, car il est très difficile d'organiser des études contrôlées contre placebo, ce qui explique la faible prévalence du syndrome. Le plus souvent, des doses élevées de corticostéroïdes sont recommandées, bien que des cas d'efficacité aient été observés avec des doses relativement faibles (par exemple, l'utilisation de prednisolone à une dose inférieure à 0,5 mg/kg par jour). Aujourd'hui, la dose moyenne de prednisolone utilisée dans le syndrome de Tolosa Hunt est de 1 à 2 mg/kg par jour.
Plan de traitement approximatif:
- Méthylprednisolone (Solu-Medron 1000 en perfusion intraveineuse goutte à goutte avec 250 ml de solution isotonique de chlorure de sodium et Panangin (10,0) par jour pendant cinq jours;
- Mildronate pour la normalisation du métabolisme cellulaire, 500 mg par injection intraveineuse par jet quotidiennement pendant 10 jours;
- Neuromidine pour améliorer la transmission des impulsions le long des fibres neuromusculaires, 20 mg par voie orale trois fois par jour;
- Clonazépam pour renforcer l'effet inhibiteur sur la transmission de l'influx nerveux et la stimulation des récepteurs des benzodiazépines, 2 mg par voie orale, et/ou Trileptal 150 mg par voie orale avant le coucher.
Il est possible de prescrire un traitement prolongé par glucocorticoïdes en utilisant des doses élevées de prednisolone. [ 8 ]
La prévention
Il est impossible de prévenir l'apparition du syndrome de Tolosa Hunt. Cela est dû, notamment, au fait que les causes de la maladie n'ont pas encore été clairement identifiées. Si des symptômes douloureux sont détectés, notamment des douleurs fréquentes au niveau de la région frontale et des orbites, une vision double et un affaiblissement des muscles oculaires, il est conseillé de consulter rapidement un spécialiste afin d'établir un diagnostic complet.
La prévention secondaire vise à prévenir les rechutes chez les patients atteints d'un syndrome de Tolosa-Hunt déjà diagnostiqué. Les points importants des mesures préventives sont les suivants:
- consultations médicales régulières, procédures de diagnostic et suivi ambulatoire spécialisé;
- traitements périodiques par corticothérapie;
- renforcer et maintenir un état adéquat du système immunitaire.
Tous ceux qui sont malades doivent essayer d’éviter les situations stressantes et traiter rapidement tout processus inflammatoire dans le corps.
Prévoir
Le pronostic du syndrome de Tolosa Hunt est jugé favorable. La corticothérapie est efficace et les cas de rémission spontanée sont fréquents, bien que certains patients présentent des effets résiduels tels qu'une altération de la fonction des muscles oculaires endommagés. En l'absence de traitement, la maladie devient récurrente. Chez les patients traités, des rechutes sont observées dans environ 35 % des cas. [ 9 ]
Une fois le traitement terminé, la capacité de travail est généralement rétablie. Cependant, cela s'applique à une maladie correctement diagnostiquée, et non à d'autres pathologies se développant sous le « masque » du syndrome. [ 10 ]
L'invalidité n'est observée que dans de rares cas. Seules des exacerbations fréquentes et documentées permettent d'attribuer le troisième groupe d'invalidité. Dans les cas difficiles, le patient est transféré vers un travail léger, non accompagné de contraintes visuelles. Si le syndrome de Tolosa Hunt est persistant et récurrent, la conduite automobile est déconseillée, en raison d'une altération de la motricité oculaire et d'une diplopie.