Expert médical de l'article

Nouvelles publications
Syndrome de Dejerine
Last reviewed: 04.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.
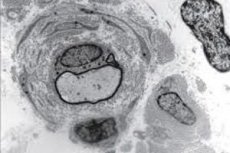
Le syndrome de Dejerine est une maladie assez rare. Il repose sur une prédisposition génétique. On l'appelle aussi neuropathie hypertrophique. On peut affirmer d'emblée que cette maladie est incurable, car toutes les maladies causées par diverses mutations et modifications génétiques sont incurables.
La première description de la maladie est due au neurologue français Dejerine, qui a initialement supposé que la maladie avait des racines génétiques profondes. Il a constaté que la maladie se transmettait de génération en génération, observée au sein d'une même famille. Il a également mené des études expérimentales qui lui ont permis de conclure que des gènes dominants étaient responsables de la transmission de la maladie. Ainsi, le conseil génétique permet de prédire si un enfant naîtra en bonne santé ou s'il développera le syndrome de Dejerine.
Malheureusement, il n'existe aucun moyen de prévenir son développement. Si la maladie est transmise à l'enfant, elle se développera inévitablement.
 [ 1 ]
[ 1 ]
Épidémiologie
Il existe actuellement de nombreux types de syndromes de Dejerine. Cependant, ils présentent tous des caractéristiques similaires: ils se manifestent entre la naissance et 7 ans. Environ 20 % des cas se déclarent au cours de la première année de vie. Au cours de la deuxième année, la maladie se déclare dans 16 % des cas.
Le syndrome de Dejerine-Sottas est le plus fréquent. Il est observé dans environ 43 % des cas. Dans environ 96 % des cas, il entraîne une invalidité complète, la personne étant confinée à un fauteuil roulant.
Le syndrome de Dejerine-Klumpke arrive en deuxième position, avec une incidence d'environ 31 % des cas. Le troisième rang est occupé par le syndrome de Dejerine-Russo, dont la fréquence d'apparition est d'environ 21 %. Ce syndrome se caractérise par l'apparition de symptômes stables en un an chez les patients ayant subi un accident vasculaire cérébral (AVC) ou un autre accident vasculaire cérébral aigu.
Le syndrome douloureux évolue de manière inégale. Chez environ 50 % des patients, la douleur survient dans le mois suivant l'AVC, chez 37 % entre un mois et deux ans, et dans 11 % des cas après deux ans. Des paresthésies et une allodynie sont observées chez 71 % des patients.
Causes Syndrome de Dejerine
La principale cause du syndrome de Dejerine est une mutation génétique transmise selon le mode autosomique. Cependant, de nombreux facteurs génétiques peuvent influencer le développement de la pathologie. Ils affectent la personne et son cerveau. Les principales causes de la maladie sont:
- traumatisme, lésion ou autres effets négatifs. Ceci est particulièrement vrai pour les nerfs crâniens. La maladie peut également être la conséquence de commotions cérébrales;
- fractures des os situés à la base du crâne;
- Inflammation des méninges, qui se manifeste de manière aiguë. L'inflammation peut avoir diverses origines. Elle peut être causée par des agents infectieux, une inflammation ou une réaction allergique. Le développement du syndrome peut également être la conséquence d'un traumatisme.
- inflammation des méninges d’origines diverses devenue chronique;
- augmentation de la pression intracrânienne.
Facteurs de risque
Certains facteurs de risque peuvent déclencher la maladie. Les personnes exposées à ces facteurs sont plus vulnérables que les autres. Parmi les facteurs de risque figurent également certaines maladies qui accompagnent la pathologie.
Le groupe à risque comprend les patients atteints de tumeurs cérébrales. Une tumeur qui exerce une pression sur le bulbe rachidien peut être considérée comme un facteur de risque. Ce groupe comprend également divers tuberculomes, des lésions vasculaires et la sarcoïdose. Les lésions cérébrales résultent d'une pression exercée sur le cerveau. Les lésions des vaisseaux cérébraux peuvent être de différentes natures. Il s'agit principalement de lésions hémorragiques, d'embolies, de thromboses, d'anévrismes et de malformations.
Parmi les facteurs contribuant au développement du syndrome de Dejerine figurent des maladies concomitantes telles que la polyencéphalite, la sclérose en plaques et la poliomyélite. Les maladies qui s'accompagnent d'une perturbation du fonctionnement normal du cerveau et de son apport sanguin peuvent également être dangereuses. Il convient tout d'abord de se méfier des perturbations de la circulation sanguine dans le lit artériel. Le groupe à risque comprend également les patients présentant une insuffisance de l'apport sanguin au douzième nerf crânien, à son noyau, à sa boucle médiale et à sa pyramide.
La syringobulbie et la paralysie bulbaire contribuent également au développement de la maladie. Ces facteurs représentent un risque élevé, car ils se caractérisent par une progression constante.
Les tumeurs cérébelleuses de divers types peuvent également être considérées comme un facteur de risque.
Les patients présentant des anomalies congénitales cérébrales sont à risque. En cas d'exposition à des agents infectieux, toxiques ou dégénératifs, le risque de développer la maladie augmente considérablement. Des facteurs tels que les produits chimiques caustiques et les substances radioactives peuvent favoriser le développement de la pathologie. Ils peuvent également provoquer une mutation génétique. Par conséquent, les femmes exposées à des substances chimiques toxiques et vivant dans une zone à fort rayonnement peuvent être à risque. La prédisposition à la maladie augmente alors fortement.
Pathogénèse
La pathogénèse de la maladie est due à une mutation génétique. Elle contribue à la perturbation de la structure des gaines des nerfs centraux. À mesure que la maladie se développe, on observe une croissance excessive des gaines conjonctives, qui font partie du tissu nerveux. Il en résulte une hypertrophie du tissu conjonctif et un dépôt de mucus entre les connexions nerveuses. Cela entraîne un épaississement important des troncs nerveux, des racines spinales et des voies cérébelleuses. Leur forme se modifie. Des processus dégénératifs affectent le tissu nerveux et les nerfs spinaux.
Symptômes Syndrome de Dejerine
Le syndrome de Dejerine peut se manifester de manières très diverses. Il est important de comprendre qu'il existe de nombreuses variantes de cette maladie, chacune se manifestant par des symptômes totalement différents. Il est donc pertinent d'aborder les signes caractéristiques de chaque type de syndrome.
Cependant, plusieurs signes précoces peuvent généralement indiquer la probabilité qu'un enfant développe une pathologie. Aux premiers stades, différents types de pathologie peuvent présenter de nombreuses similitudes.
Premiers signes
Dans la plupart des cas, la maladie se manifeste pleinement dès l'âge préscolaire. Cependant, ses premiers signes peuvent être suspectés dès la naissance. Un développement plus lent que celui de ses camarades peut être un premier signe alarmant. Il est nécessaire d'accorder une attention particulière à l'enfant qui ne s'assoit pas à l'âge approprié, qui fait ses premiers pas tardivement ou qui commence à se déplacer seul.
L'apparence de l'enfant peut également en dire long. Généralement, les muscles du visage s'affaissent. Les bras et les jambes commencent progressivement à se déformer. Ils deviennent moins sensibles et ne réagissent pratiquement plus à rien. Cet état peut s'aggraver constamment, jusqu'à l'atrophie musculaire.
Dès que l'enfant commence à se développer anormalement, il est nécessaire de consulter un médecin. Une consultation avec un neurologue est indispensable.
Lors de l'examen, le médecin découvre d'autres signes évocateurs du syndrome. On observe des contractions musculaires fibrillaires. De nombreux réflexes tendineux sont absents. Les pupilles peuvent être contractées et, dans la plupart des cas, ne réagissent pas à la lumière. Le médecin confirme des signes d'affaiblissement des muscles faciaux.
Étapes
Il existe des stades légers (initials), modérés et sévères. Le stade initial correspond à l'apparition des premiers signes de la maladie. Ce stade survient généralement pendant la petite enfance.
Le stade intermédiaire se caractérise par un retard prononcé du développement de la parole et de la motricité, divers troubles moteurs, une sensibilité altérée, une perte de certains réflexes et des réactions visuelles altérées.
Stade sévère – surdité neurosensorielle, déformations squelettiques, troubles du tonus musculaire, nystagmus. Progression de la maladie. Fin en invalidité.
Formes
Il existe de nombreuses variantes du syndrome de Dejerine, selon le type et la gravité de la lésion. Les plus fréquentes sont le syndrome alternant, le syndrome de Dejerine-Sotta, le syndrome de Dejerine-Klumpke et le syndrome de Dejerine-Rousset.
[ 21 ]
Syndrome alternant de Dejerine
Si un enfant est atteint du syndrome alternant, la langue est la première à être paralysée. De plus, la langue n'est pas entièrement touchée, mais seulement une partie. Une hémiparésie se développe du côté opposé. La sensibilité aux vibrations atteint les couches profondes. L'enfant ne distingue pratiquement pas les sensations tactiles. La cause est une thrombose ou une occlusion de l'artère basilaire, ce qui perturbe l'innervation et l'apport sanguin au bulbe rachidien.
Syndrome de Dejerine Klumpke
Dans le syndrome de Dejerine-Klumpke, les branches inférieures de l'articulation de l'épaule sont paralysées. Le membre n'est pas paralysé dans son intégralité, mais seulement partiellement. Une parésie et une paralysie des mains se développent progressivement. La sensibilité des zones concernées est fortement réduite. L'état des vaisseaux sanguins se modifie. Les réactions pupillaires sont anormales.
La paralysie s'étend progressivement aux couches profondes de la structure musculaire. Un engourdissement important est observé. Les mains s'engourdissent d'abord, puis les avant-bras et les coudes. Dans les cas graves, le nerf thoracique peut même être touché. De nombreux ptoses et myoses se développent également.
Syndrome de Dejerine-Roussy
Ce syndrome se caractérise par des lésions des artères perforantes. Sont également endommagées les zones entourant l'artère et les zones du cerveau innervées par l'artère affectée. Ce syndrome est également appelé syndrome de douleur chronique ou syndrome douloureux thalamique (post-AVC).
Ce nom s'explique par le fait que le syndrome s'accompagne d'une douleur intense, constante et perçante. La douleur est souvent insupportable. La maladie s'accompagne également d'une sensation de courbatures et de contorsion dans tout le corps. Une hyperpathie est également observée, entraînant un tonus excessif de certains muscles. Cependant, la sensibilité est fortement réduite. La maladie se caractérise également par des crises de panique, des pleurs, des cris ou des rires anormaux.
Dans ce cas, les lésions sont principalement limitées à un seul côté, qu'il s'agisse d'une jambe ou d'un bras. Dans les zones touchées, on observe principalement une douleur intense et une sensation de brûlure. La douleur épuise le patient et peut être aggravée par divers facteurs, notamment les émotions positives et négatives. Elle peut également être aggravée par la chaleur, le froid et divers mouvements.
La maladie est souvent difficile à différencier et à séparer d'autres affections. Elle présente de nombreux signes similaires à ceux d'autres lésions névralgiques. Parfois, elle ne peut être définitivement diagnostiquée qu'une fois le syndrome douloureux pleinement développé.
Syndrome de Dejerine Sottas
Le syndrome de Dejerine-Sotta est une maladie génétique. Au cours de cette maladie, l'épaisseur des nerfs centraux est altérée. Le diagnostic peut être posé dès les premiers stades de la grossesse grâce à un conseil génétique. À la naissance, l'enfant ne diffère pas d'un enfant en bonne santé. Puis, à mesure qu'il grandit, on remarque un ralentissement du développement. Ses mouvements sont faibles et il n'arrive pas à parler. Ses muscles sont très relâchés, l'enfant ne peut pas tenir sa tête, son cou ni son corps. Ses réactions visuelles sont altérées. L'enfant accuse un retard de développement important par rapport à ses pairs. Sa sensibilité diminue progressivement, ses muscles s'atrophient progressivement. Le développement n'est pas complet. Progressivement, l'atrophie atteint le système squelettique, entraînant un handicap.
Syndrome de Neri Dejerine
Dans le syndrome de Neri-Dejerine, les racines postérieures de la moelle épinière sont constamment irritées. La cause est l'ostéochondrose, une tumeur qui affecte le cerveau et le comprime. Les hernies, les pincements et les blessures contribuent également à la pression exercée sur les racines. De plus, cela peut être dû à de fortes excroissances osseuses. La principale manifestation est une douleur intense à l'endroit où s'exerce la pression sur le cerveau et ses racines.
Dans la plupart des cas, ce syndrome n'est pas le principal, mais un syndrome concomitant à diverses autres pathologies et maladies. Par exemple, il accompagne traditionnellement l'ostéochondrose. Il se caractérise par une douleur aiguë dans la région lombaire, ainsi que par une douleur lancinante au niveau du cou et de la tête, empêchant la personne de lever complètement la tête. Progressivement, cette zone durcit et la sensibilité diminue. Des spasmes musculaires sont également observés. Les membres subissent progressivement des modifications pathologiques.
Syndrome de Landouzy-Dejerine
Le terme « myopathie » est synonyme de cette maladie. Le nom de cette maladie désigne un affaiblissement musculaire en constante progression. Parallèlement, on observe le développement de diverses pathologies musculaires et des processus dystrophiques. Il ne s'agit donc pas d'une maladie distincte, mais d'un groupe de maladies. Les épaules, l'omoplate et le visage sont touchés. Il s'agit d'une pathologie génétique, transmise de génération en génération.
Elle se développe en plusieurs stades. Au premier stade, une faiblesse faciale apparaît, entraînant non seulement un affaiblissement des muscles faciaux, mais aussi une perte de forme et une déformation. Le visage acquiert alors des traits irréguliers et déformés. Le plus souvent, la maladie se reconnaît à une bouche ronde et à des lèvres inférieures et supérieures tombantes.
Progressivement, la maladie progresse à tel point que la personne ne peut plus fermer la bouche. Elle la garde ouverte d'abord pendant son sommeil, puis même pendant la journée. Progressivement, une faiblesse musculaire affecte les muscles de la ceinture scapulaire.
Dans de rares cas, les muscles pharyngés et la langue peuvent s'affaiblir. Cependant, ce symptôme n'a aucune valeur diagnostique et n'est pas aussi prononcé que d'autres.
Au stade le plus grave, la personne développe une faiblesse des muscles squelettiques. Les bras s'affaiblissent d'abord, puis les jambes. Le pronostic est décevant: invalidité.
Diagnostics Syndrome de Dejerine
Le syndrome de Dejerine peut être diagnostiqué sur la base des symptômes et des manifestations cliniques caractéristiques de la maladie. Dans certains cas, le tableau clinique est si prononcé que la maladie peut être suspectée dès l'examen clinique. Mais en réalité, la situation est bien plus complexe. D'autres maladies neurologiques peuvent se manifester de manière similaire. Il est donc important d'analyser immédiatement les signes cliniques existants et les données des examens subjectifs et objectifs. La conclusion finale repose sur des analyses de laboratoire et instrumentales. Il est également nécessaire d'étudier les antécédents médicaux et familiaux.
Tests
Le diagnostic du syndrome de Dejerine peut être confirmé par l'analyse du liquide céphalorachidien et la biopsie. L'analyse du liquide céphalorachidien permet de détecter un grand nombre de protéines et de fragments de protéines. Ces éléments constituent le signe distinctif du syndrome de Dejerine.
Dans certains cas, cela suffit à établir un diagnostic précis. Mais des doutes peuvent surgir. Par exemple, la présence de fragments protéiques dans le liquide céphalorachidien (LCR) peut indiquer d'autres maladies neurologiques. Par conséquent, en cas de doute, une biopsie est réalisée. Elle nécessite le prélèvement de fibres nerveuses au niveau des muscles du coude et du mollet. Le syndrome de Dejerine est précisément indiqué par la présence d'une hypertrophie du tissu nerveux. Dans cette pathologie, les gaines des fibres nerveuses s'épaississent fortement.
De plus, l'étude des tissus nerveux au microscope révèle non seulement un épaississement des membranes, mais aussi une diminution significative du nombre de fibres. Une déminéralisation se produit également, réduisant ainsi le nombre de fibres nerveuses.
Diagnostic instrumental
Mais il arrive parfois que même les tests ne suffisent pas à confirmer avec certitude le syndrome de Dejerine. Un équipement spécifique est alors nécessaire. Dans la plupart des cas, on utilise l'imagerie par ordinateur et l'imagerie par résonance magnétique. Ces méthodes permettent de détecter le degré de lésion du douzième nerf. Après réception des résultats, le médecin choisit un traitement. Ce traitement est prescrit par un neurologue ou un neurochirurgien.
Diagnostic différentiel
L'essence du diagnostic différentiel est de distinguer clairement les symptômes d'une maladie de ceux d'une autre présentant des manifestations similaires. Dans le syndrome de Dejerine, cet aspect est essentiel, car le pronostic et le traitement ultérieurs en dépendent. Cette maladie peut souvent être confondue avec d'autres maladies neurologiques, comme la paralysie.
Une fois le diagnostic confirmé, il est nécessaire de le différencier des autres formes, c'est-à-dire de déterminer clairement à quel type spécifique de syndrome de Dejerine nous avons affaire. Une biopsie et une analyse du liquide céphalorachidien peuvent y contribuer.
La présence d'un syndrome de Dejerine est indiquée par la présence de protéines dans le liquide céphalorachidien et l'épaississement des fibres nerveuses à la biopsie. Le type de syndrome est généralement déterminé par le tableau clinique et les signes pathognomoniques, c'est-à-dire les signes caractéristiques d'une maladie particulière, qui lui sont propres et qui en constituent le signe distinctif.
Qui contacter?
Traitement Syndrome de Dejerine
La maladie étant génétique, il est important de comprendre qu'il sera impossible de la guérir complètement et de s'en débarrasser. Il est fort probable qu'elle progresse et qu'il soit impossible de l'arrêter. Cela ne signifie pas pour autant qu'il ne vaut pas la peine de la traiter. Il suffit de choisir le traitement le plus soigneusement et le plus rationnellement possible. Il peut ralentir la progression de la maladie et soulager considérablement les souffrances du patient.
Lors du choix du traitement, ils sont guidés par le fait que la thérapie étiologique est irrationnelle. La thérapie étiologique est définie comme un traitement visant à éliminer la cause. Pour une maladie génétique, il est impossible d'éliminer la cause. Par conséquent, le traitement symptomatique demeure, c'est-à-dire un traitement visant à soulager les symptômes de la maladie, à améliorer l'état général et à assurer le bien-être du patient. Les schémas thérapeutiques peuvent être très variés. Tout dépend du symptôme prédominant et de ce qui inquiète le patient à ce moment précis. Afin de soulager les symptômes et de prévenir la progression de la maladie, une thérapie complexe est utilisée.
Le traitement complexe comprend généralement des analgésiques, car le syndrome s'accompagne presque toujours de sensations douloureuses. En l'absence de douleur (ce qui est assez rare), ces médicaments peuvent être exclus.
Une thérapie métabolique, qui améliore les processus métaboliques, favorise une meilleure nutrition des tissus et élimine les métabolites, est indispensable. Ce type de traitement vise principalement à préserver le tissu musculaire, très sensible aux processus dégénératifs et susceptible de s'atrophier. L'objectif principal de cette thérapie est de prévenir l'atrophie.
Il est également nécessaire d'utiliser des médicaments pour améliorer la conductivité nerveuse. Ils permettent de normaliser les processus métaboliques dans le tissu nerveux, de restaurer ou de maintenir la sensibilité des terminaisons nerveuses et de prévenir la mort des récepteurs.
Outre le traitement médicamenteux, une physiothérapie peut être prescrite. Des séances de massage, de thérapie manuelle et diverses thérapies alternatives peuvent être nécessaires. Il existe actuellement de nombreux produits orthopédiques permettant de prévenir le développement de maladies osseuses. Il est également possible de prévenir les déformations du pied. Les contractures articulaires peuvent également être prévenues grâce à des produits orthopédiques.
Parfois, le traitement vise à éliminer la cause de la maladie. Bien sûr, il ne s'agit pas d'une cause génétique. Par exemple, dans certains cas, malgré une prédisposition génétique, la maladie ne se manifeste pas. Cependant, sous l'effet d'un facteur, la maladie commence à se développer ou à progresser. La cause peut alors être une thrombose artérielle. L'artère endommagée comprime la partie adjacente du cerveau, perturbant son apport sanguin. Dans ce cas, il est conseillé de tenter d'éliminer la cause: retirer la thrombose. Une intervention chirurgicale peut alors être nécessaire.
Dans d’autres cas, une thérapie de soutien continue est nécessaire.
Médicaments
Les médicaments sont utilisés exclusivement pour soulager les symptômes. Par exemple, pour traiter la douleur, il est recommandé d'utiliser du cabrazépam à une dose de 3 à 5 mg/kg de poids corporel, 2 à 3 fois par jour.
Le kétorolac peut également être recommandé à une concentration de 60 mg/jour, 2 fois par jour. Le trométamol est utilisé à 60 mg/jour, 2 fois par jour, et le cétonal à 50 mg, 1 à 2 fois par jour. La dose quotidienne maximale est de 100 à 150 mg.
Vitamines
Les vitamines sont nécessaires au maintien de l'état général de l'organisme, au renforcement du système immunitaire et à la protection contre les maladies infectieuses et autres. Elles contribuent également au bien-être général de l'enfant. Voici les principales vitamines nécessaires au bon fonctionnement de l'organisme (dose quotidienne):
- B – 2-3 mg
- PP – 30 mg
- H – 7-8 mcg
- C – 250 mg
- D – 20 mcg
- E – 20 mg.
Traitement de physiothérapie
La physiothérapie ne peut pas guérir le syndrome de Dejerine. Cependant, dans certains cas, elle peut être utilisée. Elle est généralement utilisée dans un but précis. Ainsi, l'électrophorèse augmente significativement la perméabilité tissulaire et permet aux médicaments de pénétrer les tissus plus rapidement et plus efficacement. Certaines procédures de physiothérapie peuvent réduire la douleur, détendre les muscles et soulager les spasmes. Elles peuvent améliorer significativement l'état général du corps. La physiothérapie contribue également à soulager la douleur.
Remèdes populaires
Il existe des remèdes populaires pour soulager le patient. Il est impossible de guérir le syndrome de Dejerine, mais cela ne signifie pas qu'il faille abandonner et ne rien faire pour soulager ou atténuer les symptômes. Les remèdes populaires aident à surmonter les symptômes et à soulager la douleur. Ils apportent un soutien psychologique important au patient. La maladie est incurable, mais sa progression peut être ralentie.
En cas de parésie, de paralysie ou d'affaiblissement des muscles faciaux, il est recommandé de consommer de l'avoine. Consommez du jus d'avoine verte. Prenez un tiers de verre deux fois par jour. Il est préférable de le prendre avant les repas. L'effet est un renforcement général.
La paralysie et la parésie sont soulagées par la menthe et la mélisse. Il est recommandé de les infuser, de les préparer en décoction et de les boire chaudes. Ces plantes sont relativement sûres; on peut donc les utiliser en grande quantité, sans toutefois les abuser. Environ un litre par jour est autorisé. On peut également les ajouter à du thé selon son goût. Buvez-les en quantité illimitée, selon vos envies et votre humeur. Elles favorisent généralement la détente musculaire, normalisent le système nerveux et ont un effet calmant.
La menthe et la mélisse peuvent également être mélangées au gui, à parts égales, et utilisées en décoction. Dans ce cas, il est conseillé de les utiliser en quantité limitée, environ 20 à 30 ml trois fois par jour. Cette infusion aide à soulager les spasmes et les douleurs, à détendre les muscles et à calmer le système nerveux.
Les bains aux herbes médicinales ont un effet bénéfique sur le corps. Vous pouvez préparer un bain à partir de ces plantes. Pour cela, préparez séparément environ 2 à 3 litres d'infusion forte, puis versez-la dans un bain à température agréable. Il est recommandé de prendre des bains de 20 à 30 minutes. Ils tonifient les muscles et normalisent l'activité du système nerveux. Les herbes peuvent être combinées et alternées. Vous pouvez utiliser une décoction de conifères: pin, sapin, cèdre. Vous pouvez ajouter de la camomille, du tilleul, de la framboise et de l'ortie.
[ 31 ], [ 32 ], [ 33 ], [ 34 ]
Traitement à base de plantes
En cas de troubles de la coordination, de parésie ou de paralysie, l'éphédra peut être utilisée. Elle s'utilise sous forme de décoction. Infusez environ 5 g de plante dans 500 ml d'eau bouillante. Vous pouvez en boire 2 à 3 cuillères à soupe trois fois par jour.
Pour normaliser l'état musculaire, apaiser, soulager les spasmes et la douleur, utilisez une décoction ou une infusion de valériane. La teinture alcoolique de valériane est disponible dans le commerce. Le mode d'administration est généralement indiqué sur l'emballage.
Pour préparer une décoction maison, versez environ 5 g de la plante dans un verre d'eau et buvez-la tout au long de la journée. Vous pouvez également l'ajouter à votre thé.
La décoction de camomille peut être utilisée de la même manière. Elle possède également un effet anti-inflammatoire et normalise le système immunitaire et le métabolisme.
Il est recommandé d'utiliser une cuillère à soupe de décoction de calendula trois fois par jour. Elle a un effet anti-inflammatoire et soulage les gonflements.
Homéopathie
Les remèdes homéopathiques peuvent également avoir un effet positif, améliorer l'état général de l'organisme et soulager certains symptômes. Les effets secondaires sont rares si la posologie et le mode d'administration sont correctement respectés. Il est important de noter que de nombreuses substances ont un effet cumulatif, ce qui signifie que l'effet n'apparaît qu'après la fin du traitement complet, ou après un certain temps. Il est nécessaire de respecter les précautions de base: consultez un médecin avant de prendre ce médicament, car certaines substances peuvent être incompatibles entre elles ou avec les médicaments. Les conséquences peuvent être imprévisibles.
En cas de paralysie flasque, de parésie, d'hyperkinésie et d'affaiblissement des muscles faciaux, il est recommandé de prendre Securinega sibirica. Versez environ 15 g de feuilles et de petites branches broyées dans 250 ml d'eau bouillante. Laissez infuser jusqu'à refroidissement. Filtrez et prenez un tiers de verre, deux fois par jour.
- Collection n° 1. Pour les lésions de la moelle allongée, du cervelet
Prenez des feuilles de framboisier, de cassis, de valériane et d'agripaume dans un rapport de 2:1:2:1. Prenez-en une infusion, un tiers de verre trois fois par jour.
- Collection n° 2. Pour la paralysie spastique
Prenez des feuilles de mélisse, de camomille, de sauge et de menthe dans un rapport de 1:1:2:2. Prenez-en une infusion trois fois par jour, un tiers de verre.
- Collection n° 3. Pour le syndrome douloureux, les spasmes, la paralysie
Prenez des feuilles de stévia, de l'ortie, des cônes de houblon et des fleurs d'échinacée dans un rapport de 2:1:1:1. Prenez-les en infusion trois fois par jour.
Traitement chirurgical
Dans certains cas, seule une intervention chirurgicale peut être efficace. Par exemple, si le patient présente une blessure ou une tumeur, celle-ci doit être retirée. Une thrombose ou une occlusion artérielle peut également nécessiter une intervention chirurgicale.
En cas de pathologie des vaisseaux sanguins, une intervention chirurgicale intravasculaire mini-invasive est efficace.
Si une artère spécifique est affectée, une intervention chirurgicale peut être nécessaire pour améliorer la circulation cérébrale et normaliser l’innervation de cette zone.
Mais dans certains cas, certaines pathologies ne peuvent être opérées. Il peut s'agir de diverses anomalies congénitales ou de blessures.
Prévoir
L'évolution de la maladie est toujours progressive, les périodes de rémission étant courtes. Le pronostic est défavorable. Cela est principalement dû au fait que les principaux processus dégénératifs touchent le système nerveux et le cerveau. À mesure que la maladie progresse, la capacité de travail diminue. Le patient est finalement confiné à un fauteuil roulant ou au lit.
[ 39 ]