Nouvelles publications
De nouvelles découvertes permettent de mieux comprendre les causes du syndrome de Rett
Dernière revue: 02.07.2025

Tout le contenu iLive fait l'objet d'un examen médical ou d'une vérification des faits pour assurer autant que possible l'exactitude factuelle.
Nous appliquons des directives strictes en matière d’approvisionnement et ne proposons que des liens vers des sites de médias réputés, des instituts de recherche universitaires et, dans la mesure du possible, des études évaluées par des pairs sur le plan médical. Notez que les nombres entre parenthèses ([1], [2], etc.) sont des liens cliquables vers ces études.
Si vous estimez qu'un contenu quelconque de notre contenu est inexact, obsolète ou discutable, veuillez le sélectionner et appuyer sur Ctrl + Entrée.

Le syndrome de Rett est un trouble neurodéveloppemental rare pour lequel il n'existe actuellement aucun remède ni traitement efficace. Il provoque de graves symptômes physiques et cognitifs, dont beaucoup se recoupent avec les troubles du spectre autistique.
Le syndrome de Rett est causé par des mutations du gène MECP2, fortement exprimé dans le cerveau et qui semble jouer un rôle important dans le maintien de la santé des neurones. Ce gène est situé sur le chromosome X et le syndrome touche principalement les filles. Afin de développer des traitements contre le syndrome de Rett, les chercheurs souhaitent mieux comprendre le gène MECP2 et ses fonctions cérébrales.
Les chercheurs, dont Rudolf Jaenisch, cofondateur de l'Institut Whitehead, étudient le gène MECP2 depuis des décennies, mais de nombreux éléments fondamentaux le concernant restaient inconnus. La protéine codée par ce gène, MECP2, intervient dans la régulation des gènes; elle se lie à l'ADN et influence l'expression de divers autres gènes, ou la quantité de protéines qu'ils produisent.
Cependant, les chercheurs ne disposaient pas d’une liste complète des gènes affectés par MECP2, et il n’y avait pas de consensus sur la manière dont MECP2 affecte ces gènes.
Les premières études sur MECP2 suggéraient qu'il s'agissait d'un répresseur réduisant l'expression de ses gènes cibles, mais des recherches menées par Jaenisch et d'autres avaient précédemment montré que MECP2 agissait également comme activateur, augmentant l'expression de ses gènes cibles – et qu'il pourrait même être un activateur. On ignorait également le mécanisme d'action de MECP2, ou comment la protéine induit précisément des modifications de l'expression génétique.
Les limites technologiques ont empêché les chercheurs de répondre à ces questions. Mais Yanish, Yi Liu, postdoctorant au sein de son laboratoire, et Anthony Flamier, ancien membre de son laboratoire et aujourd'hui professeur adjoint au centre de recherche du CHU Sainte-Justine de l'Université de Montréal, ont utilisé des techniques de pointe pour répondre à ces questions en suspens sur le gène MECP2 et mieux comprendre son rôle dans la santé et les maladies cérébrales.
Leurs résultats ont été publiés dans la revue Neuron et les chercheurs ont également créé un référentiel en ligne de leurs données MECP2, le portail MECP2-NeuroAtlas, comme ressource pour d'autres chercheurs.
« Je pense que cette étude va fondamentalement changer notre compréhension de la façon dont MECP2 provoque le syndrome de Rett. Nous avons une compréhension totalement nouvelle du mécanisme, et cela pourrait ouvrir de nouvelles perspectives pour le développement de traitements contre la maladie », déclare Janisch, également professeur de biologie au MIT.
Une meilleure compréhension du gène MECP2 dans le cerveau
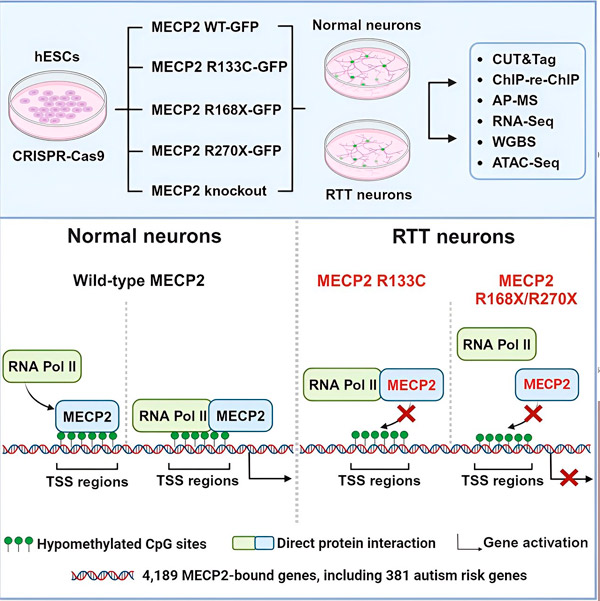
Les chercheurs ont d'abord établi une carte détaillée des sites de liaison de MECP2 dans les séquences génétiques neuronales humaines, soit à l'intérieur des gènes, soit dans les régions régulatrices de l'ADN proches. Ils ont utilisé une approche appelée CUT&Tag, qui permet d'identifier avec une grande précision les interactions entre les protéines et l'ADN.
Les chercheurs ont identifié plus de 4 000 gènes associés à MECP2. Ils ont répété leur cartographie dans des neurones présentant des mutations courantes du gène MECP2 associées au syndrome de Rett afin de déterminer où le gène MECP2 est déficient dans la maladie.
En connaissant les gènes auxquels se lie MECP2, Liu et Flamier ont commencé à établir des liens entre les cibles de MECP2 et la santé cérébrale. Ils ont découvert que nombre de ses cibles sont impliquées dans le développement et le fonctionnement des axones et des synapses neuronales.
Ils ont également comparé leur liste de cibles MECP2 avec la base de données de gènes associés à l'autisme de la Simons Foundation Autism Research Initiative (SFARI) et ont découvert que 381 gènes de cette base de données sont des cibles MECP2.
Source: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Ces résultats peuvent aider à clarifier les mécanismes sous-jacents aux symptômes de l’autisme dans le syndrome de Rett et fournir un bon point de départ pour étudier le rôle possible du MECP2 dans l’autisme.
« Nous avons créé la première carte intégrée de l'épigénome MECP2 dans les maladies et la santé, et cette carte pourra orienter les recherches futures », explique Liu. « Connaître les gènes cibles de MECP2 et ceux directement perturbés dans la maladie constitue une base solide pour comprendre le syndrome de Rett et s'interroger sur la régulation des gènes dans les neurones. »
Les chercheurs ont également examiné si MECP2 augmentait ou diminuait l'expression de ses gènes cibles. Conformément à l'histoire de MECP2, identifié par certains comme activateur et par d'autres comme répresseur, Liu et Flamier ont trouvé des exemples où MECP2 jouait les deux rôles.
Cependant, alors que MECP2 est plus souvent considéré comme un répresseur, Liu et Flamier ont découvert qu'il s'agit principalement d'un activateur, confirmant ainsi les résultats précédents de Jaenisch et Liu. Une nouvelle expérience a montré que MECP2 active au moins 80 % de ses cibles, et une autre a révélé qu'il active jusqu'à 88 % de ses cibles.
La carte des gènes cibles créée par les chercheurs a apporté des informations supplémentaires sur le rôle activateur de MECP2. Ils ont constaté que les gènes activés par MECP2 se lient généralement à une région de l'ADN en amont du gène, appelée site d'initiation de la transcription.
C'est à ce site que la machinerie cellulaire initie le processus de transcription d'un gène en ARN, après quoi l'ARN est traduit en une protéine fonctionnelle, fruit de l'expression génique. La présence de MECP2 au site d'initiation de la transcription, où débute l'expression génique, est cohérente avec son rôle d'activateur génique.
Les chercheurs ont ensuite cherché à déterminer le rôle de MECP2 dans l'activation des gènes. Ils ont étudié les molécules auxquelles MECP2 se lie à ce site, en plus de l'ADN, et ont découvert que MECP2 interagit directement avec un complexe protéique appelé ARN polymérase II (ARN Pol II). L'ARN Pol II est une machine cellulaire essentielle qui transcrit l'ADN en ARN. L'ARN Pol II ne peut pas trouver les gènes par elle-même; elle a donc besoin de divers cofacteurs, ou protéines collaboratrices, pour l'aider à accomplir sa tâche.
Les chercheurs suggèrent que MECP2 joue un rôle de cofacteur, aidant l'ARN Pol II à initier la transcription au niveau des gènes où MECP2 se lie. L'analyse structurale de MECP2 a permis d'identifier les parties de la molécule qui se lient à l'ARN Pol II, et d'autres expériences ont confirmé que la perte de MECP2 réduit la présence de l'ARN Pol II aux sites de démarrage de la transcription appropriés, ainsi que les niveaux d'expression des gènes cibles.
Cela suggère que le syndrome de Rett pourrait être causé par une diminution de la transcription des gènes ciblés par MECP2, due à des mutations de MECP2 l'empêchant de se lier à l'ARN Pol II ou à l'ADN. Conformément à cette idée, les mutations de MECP2 les plus courantes associées à la maladie sont des troncatures: des mutations dans lesquelles une partie de la protéine est manquante, ce qui peut altérer l'interaction entre MECP2 et l'ARN Pol II.
Les chercheurs espèrent que leurs découvertes changeront non seulement notre compréhension du MECP2, mais qu’une compréhension plus approfondie et plus large de la façon dont le MECP2 influence le développement et le fonctionnement du cerveau pourrait conduire à de nouvelles perspectives qui aideront les personnes atteintes du syndrome de Rett et de troubles associés, y compris l’autisme.
« Ce projet illustre parfaitement la nature collaborative du laboratoire Janisch », déclare Flamier. « Rudolf et moi avions un problème spécifique lié au syndrome de Rett, et j'avais une expérience de la technologie CUT&Tag, qui pouvait le résoudre. Grâce à nos échanges, nous avons compris que nous pouvions combiner nos efforts, et nous disposons désormais d'une importante base de données sur MECP2 et ses liens avec la maladie. »